Abstract
For more than a decade, the nuclear factor kappa-B (NFκB) family and their signaling pathways have proved crucial to the immune system's functioning. According to research, this factor is implicated in almost every immune system event, including immune cell development and function, activation, and pathogen-activated cell death. A considerable body of evidence suggests the idea that, in addition to being a possible transcription factor, it plays a very significant role in the establishment of inflammation-associated immunological responses in host cells. Inflammation is a cascade of events that involve vasodilation and immune cell migration to the site of infection in order to defend the host. However, if dysregulation occurs, it may lead to the emergence of chronic inflammatory illnesses. When immune cells migrate to an inflamed area NFκB activation occurs, which causes release of pro-inflammatory cytokines and development of inflammatory response. Furthermore, this complex transcription factor activation takes part in several innate and adaptive immunity-related cellular processes, including immune cell proliferation and differentiation. Grasp the relationship between the immune system and inflammation requires a thorough understanding of NFκB's capabilities. This review explores the role of NFκB activity as a key inflammatory mediator in various malignancies and auto-inflammatory diseases. We have shed light on how NFκB regulates inflammatory responses by producing cytokines and chemokines and directing the transcription of inflammatory molecules involved in both innate as well as adaptive immunity.
Keywords
Inflammation, NFκB, Cytokines, Innate immunity, Adaptive immunity, Treg, PAMP, TLR, Tumor-microenvironment, Immuno-suppression, Cancer, Autoimmunity
Abbreviations
NFkB: Nuclear Factor Kappa B; PAMP: Pathogen-Associated Molecular Pattern; PRR: Pattern Recognition Receptor; TNFR: Tumor Necrosis Factor Receptor; IKK: IκB Kinase; BAFF: B Cell Activating Factor; LTbR: lymphotoxin Beta Receptor; RANK: Receptor Activator of Nuclear Factor Kappa B; NIK: NFkB-Inducing Kinase; TLR: Toll-Like Receptor; NLR: Nod-Like Receptor; MyD88: Myeloid Differentiation Primary Response-88; MHC: Major Histocompatibility Complex; TCR: T Cell Receptor; IFNg: Interferon Gamma; GATA3: GATA Binding Protein-3; RORgt: RAR-Related Orphan Receptor; BCR: B Cell Receptor; Breg: B-Regulatory Cell; IL10: Interleukin-10; CXCL2: Chemokine (C-X-C motif) Ligand-2; CAC: Colitis-Associated Cancer; TAM: Tumor-Associated Macrophage; VEGF: Vascular Endothelial Growth Factor; Treg: T-Regulatory Cell; TME: Tumor Microenvironment.
Introduction
The immune components play a critical role in defending multicellular organisms against infections [1]. To accomplish this feat, it has created naturally-hardwired defense system, including primary and secondary lymphoid organs for the generation and selection of immune cells, distinct immune cells for specific functions, and inflammation to alarm the organism's whole physiology [2]. The immune system has evolved not only to eliminate pathogens, but during the training process of immune cells, it also learns to discriminate between self and non-self in order to prevent autoimmunity and maintain homeostasis through self-tolerance [3], as well as induce anti-tumor immunity by attacking transformed cells [4].
With the advancement of molecular technologies and genetically modified mouse models, scientists from all around the world are going deep into the enigma of our immune system's numerous functions. Cell signaling is involved in the immune cells' communication system [5]. Different cytokines, chemokines, and compounds such as pathogen-associated molecular patterns (PAMPs) induce distinct signaling pathways that are required for immune cells to operate properly [6]. Numerous transcription factors are engaged during this cell signaling process either stimulating or inhibiting gene transcription, resulting in their increased or decreased production of messenger RNA and, consequently, determining the level of protein products. All these plays a very important role in immune responses [7].
Nuclear factor kappa-B (NFkB) is a family of transcription factors that is activated in reaction to various immunological stimuli [8]. For several decades, researchers studying immunology have discovered a link between NFkB molecules and nearly all of the immune system's critical processes, including pathogen-activated death, activation of T cells and B cells, development of inflammation, and malignancy [9]. The NFkB family transcription factor includes five structurally similar members: NFkB1 (p50), NFkB2 (p52), RelA (p65), RelB, and c-Rel. They form homo-or heterodimers and interact with certain DNA elements to regulate target gene transcription. When NFkB is inactive, it is sequestered in the cytoplasm by inhibitory IkB family proteins [10]. The study reveals that NFkB is activated by two primary signaling routes: the canonical and non-canonical (or alternative) pathways. Although their modes of action vary, both pathways play a crucial role in controlling immunological and inflammatory reactions [11-13]. The canonical NFkB pathway can be triggered by various type of stimuli, including cytokines, pattern-recognition receptors (PRRs), TNF receptor (TNFR) superfamily, and also by T cell receptor (TCR) and B cell receptor (BCR) mediated signaling [14]. In canonical pathways, all these stimuli merge into one target for proteosomal degradation of IkB by a kinase enzyme called the IkB kinase (IKK) complex [8,15]. Degradation of IkB causes releases of p50/RelA and p50/c-Rel dimers, which results in rapid and transient nuclear translocation (Figure 1) [16].

Figure 1. Activation of NFkB signaling by diverse stimuli The canonical NFκB pathway is activated by signaling through pattern recognition receptors (TLR), TCR, BCR, TNFR and IL1R. The IKK, a kinase enzyme, targets IkB for proteosomal degradation. Degradation of IκB causes the release of p50/RelA from its inhibitory complex, leading to rapid and transient nuclear translocation and activation of NFκB-inducible genes. Ligands of TNFR superfamily members, including LTβR, BAFFR, CD40, and RANK, activate the non-canonical NFκB pathway. This pathway involves ubiquitination and processing of NFκB2 precursor protein p100 by NFκB-inducing kinase (NIK). After processing, it forms mature NFκB2 (p52) and translocates to nucleus as NFkB complex (p52/RelB) to activate its target genes. Developed in BioRender.com.
Non-canonical pathways may also be activated by a particular collection of stimuli, such as ligands of a subset of TNFR superfamily members, such as LTβR, BAFFR, CD40, and RANK. This pathway requires NFκB-inducing kinase (NIK) to ubiquitinate and process the NFκB2 precursor protein p100. After processing, it generates mature NFkB2 (p52) and translocate to the nucleus as the NFkB complex p52/RelB (Figure 1) [13,17,18]. The classical NFκB route governs nearly every facet of immune responses, but the non-canonical pathway has developed as an additional signaling axis for particular adaptive immune system activities [10,12].
Numerous studies involving NFkB inhibitors have established the significant role of this family of transcription factor in developing inflammation. Inflammation occurs when infected or injured tissue or tissue-resident mast cells or dendritic cells detect and propagate inflammatory signals. The recruitment and triggering of neutrophils, macrophages, and other effector cells as a result of this signal causes inflammation to progress [9]. Expression of adhesion molecules responsible for extravasation of leukocytes to target site is controlled by NFkB [19]. NFkB has also played very important role in regulating matrix metalloproteinases (MMPs) expression which are the primary mediators of local inflammation and leukocyte chemotaxis [20]. Meanwhile, multiple investigations have demonstrated that NFkB may have a role in the resolution of inflammation and subsequent tissue healing as blocking NFkB during the resolution phase prolongs inflammatory responses and hinders tissue regeneration, it is not recommended [21]. This failure subsequently leads to the development of chronic inflammatory response, which is the source of several common pathologies. So, considering that NFkB is involved in practically every aspect of immune responses, targeting NFkB in diverse immunological disorders such as chronic inflammatory illnesses and cancer is important for developing effective treatment procedure to combat these dreadful diseases. Here, we have covered the role of NFkB in generation and survival of leukocytes – involved in both innate as well as adaptive immunity as their role is critical during immunological responses to inflammation and also underlines the mechanism of NFkB participation in several pathological conditions.
NFkB in Innate Immunity
Innate immunity is the immune system's first line of defense in our bodies. During infection it is triggered in response to pathogen-associated molecular patterns (PAMPs) such as LPS, peptidoglycan, lipoproteins, bacterial DNA, and double-stranded RNA viruses [22]. It is recognized by the expression of pattern recognition receptors (PRRs) like Toll-like receptors (TLRs), RIG-I-like receptors (RIG-ILRs), NOD-like receptors (NLRs), C-type lectin-like receptors (CLRs), and cytosolic DNA sensors expressed by innate immune cells such as neutrophils, macrophages and dendritic cells [6,23]. Innate immunity, which is phylogenetically conserved in insects and humans, uses TLR-mediated NFkB activation to give a rapid response to pathogenic bacteria [24,25]. NFkB modulates expression of cytokines, adhesion molecules, acute phase proteins, inducible enzymes, chemokines, and antimicrobial peptides like -defensins, all of which are critical components of innate immune system to invading pathogens [26-29]. The NFkB transcription factor play critical roles in innate immune cells' formation and immuno-regulatory responses.
Pennington et al. discovered that macrophage development from monocyte progenitors is linked to IKK and NFkB activation, which protects these cells against apoptosis. They also suggested that p21 (Waf1/Cip1) is a key NFkB target gene for macrophage survival [30]. According to Lawrence et al. and Li et al. studies, reduced activity of IKK1 in macrophages results in higher NFkB activation, increased antigen presentation to T cells, and increased bacterial clearance [31,32]. The role of NFkB-signaling in controlling M1/M2 macrophage polarization, which is predominantly influenced by pathophysiological circumstances, is crucial. In inflammatory settings, M1 macrophages produce various pro-inflammatory cytokines like IL12, IL6, IL1, TNFα, chemokines, and encourage the formation of Th1 and Th17 cells. M2 macrophages, on the other hand, are known for producing anti-inflammatory cytokines like IL10 to reduce inflammation. TLR4-signaling mediated by MyD88 promotes M1 macrophage polarization, with NFkB as the main transcription factor (Figure 2a) [33-36]. In macrophages, c-Rel regulates the transcription of IL12B (IL12p40), which is an important inducer of T helper1 cell growth [37]. In bacterial sepsis, mice with impaired c-Rel and p50 NFkB proteins have macrophages that are unable to phagocytose and destroy bacteria [38]. Huang et al. stated that phosphorylation of IkB and activation of TNFα induced NFkB stimulate the production of complement factor-B (Bf), a serine protease in macrophage to activate complement pathway [39].

Figure 2. Role of NFkB in immune responses. a.NFkB activation is necessary for myeloid cell development and function, including dendritic cell maturation, macrophage polarization, and neutrophil survival. b. NFkB activation is essential for both positive and negative selection during T cell differentiation with differential TCR affinity. c. During B cell development, cells that detect self-antigens through B cell receptor (BCR) perish due to activation of pro-apoptotic genes, while cells that do not recognize self-antigens survive due to upregulation of pro-survival proteins caused by NFkB activation. Developed in BioRender.com.
Under inflammatory condition monocytes differentiate into dendritic cells (DC), which is essential for response to pathogens. Nevertheless, research has demonstrated that canonical stimuli and pathway elements are necessary for DCs to mature into professional antigen-presenting cells in a RelB-dependent manner (Figure 2a). It has been reported by Shih et al. that RelB dependent DC activation occurs through RelB-p50 dimer, which is regulated by conventional IkBs, IkBα, and IkBε, rather than the non-canonical RelB-p52 route [40]. RelB-lowering DCs are unable to stimulate antigen-specific T cell responses in vitro or in vivo [41]. It's also been discovered that p100, not IkB proteins, regulates RelB's translocation into the nucleus and that p100's inhibitory domain inhibits DC development in the precursor stage [42]. Aside from these, the NFkB-like factor c-Rel plays a function in DC cell growth. T cells cannot be successfully activated by DCs generated from bone marrow that lack c-Rel [43]. Because CD40L-CD40 signaling promotes the NFkB pathways, DCs lacking p50 and c-Rel have a lower survival rate [44,45]. DC formation has been demonstrated to be hampered by blocking upstream components of the NFkB pathway. TRAF6-deficient DCs cannot excite naïve T lymphocytes because they cannot upregulate MHCII and B7.2 surface expression or generate inflammatory cytokines. The NFkB activation regulates antigen presentation in DCs as well [46,47]. Non-immune cells like fibroblasts, endothelium, and epithelial cells also respond to pathogens by the activation of NFkB. In a TLR2-dependent manner, fibroblasts respond to necrotic cells by activating NFkB and producing chemokines [48].
The function of NFkB in neutrophils, mast cells, and eosinophils are still unknown. Neutrophils are drawn to inflamed regions to kill the pathogen. PAMPs are recognized by neutrophils, which activate the conventional NFkB-signaling pathway [49]. NFkB performs an anti-apoptotic effect and determine neutrophils survivability (Figure-2a). Studies shown that use of NFkB inhibitors promote the apoptosis of human neutrophils. Inhibition of IKK complex by using NEMO-binding domain peptide (NFkB pathway inhibitor) results in cell death in these cells [50]. NFkB activity in neutrophils is increased by several TLR ligands, allowing anti-apoptotic genes to be expressed [51]. Neutrophils lack the proteins p52 and RelB [9,52], both of which are essential for the survival of lymphocytes. NFkB thus fulfils its projected role as a pro-survival and pro-inflammatory factor in neutrophils. Neutrophils undergo apoptosis shortly after activation to reduce their inflammatory potential. Nuclear IkBa levels in neutrophils are excessively high, resulting in rapid apoptosis [53].
NFkB in Adaptive Immunity
Although innate responses can operate as a potent defense barrier on their own, the adaptive immune system must be alerted and activated for immune responses to be robust and long-lasting. Antigen-presenting cells' (APCs) development and activation play a major role in this, as they then direct T and B cells to carry out the adaptive response. Signaling through these antigen-specific B cell and T cell receptors is thus important to the adaptive immune response. NFkB exerts its role as a transcriptional regulator in the adaptive immune responses [54]. NFkB plays role as a pro-survival factor in T and B cell responses, as well as the control of genes involved in effector cell growth. Activated NFkB control the resolution of the response and encourages the establishment of memory response after the pathogen has been cleared.
The fact that most members of the NFkB family are expressed in T cells suggests that they are engaged in T cell activities. After TCR ligation, NFκB is activated, which promotes antigen-specific lymphocyte growth and effector cell maturation. During T cell lymphopoiesis, the anti-apoptotic protein Bcl2 is induced by NFkB. In various studies, NFkB has been linked to the survival of mature lymphocytes. B cells from RelA, p100, p105, and c-Rel deficient mice exhibit increased apoptotic sensitivity and/or shorter survival in-vitro [55-57]. NFkB also regulates the costimulatory molecule B7h, which is a ligand for the T cell costimulatory protein ICOS [58]. These results suggest a role for NFκB in the regulation of accessory cell functions influencing the development of adaptive responses.
NFkB controls both positive and negative thymocyte selection. After high-affinity TCR ligation during negative selection, NFkB enhances the expression of pro-apoptotic genes [59-61]. NFkB’s role in thymocyte positive selection is aided by anti-apoptotic genes (Figure 2b). Temporal regulation of NFκB/Bcl2-axis controls the differentiation and survival of specialized subsets of T cells during antigen-dependent selection in response to T cell receptor signals.
The expression of CD40 and major histocompatibility complex class-I (MHC-I), which are necessary for the formation of CD8+ T cell responses, in adequate quantities by embryonic fibroblasts is dependent on RelA [62]. It has been demonstrated that chemical inhibitors of NFkB activation change DC maturation and boost the synthesis of MHC-II and B7 costimulatory molecules, both of which are necessary for effective CD4+ T cell responses [63]. In mature T cells, signaling through the TCR activates protein kinase Cθ, resulting in NFkB activation, which is required in TCR-mediated proliferative signals [64]. NFkB activity is needed for the protection of rapidly proliferating activated T cells from apoptosis as well as the release of cytokines like IL12 [65]. Interestingly, c-Rel-deficient T cells lack Th1 proliferation and IFNg production, implying that NFkB family members play a selective role in Th1/Th2 differentiation regardless of the innate response. Inhibition of NFkB enhances apoptosis of activated T cells [66-69]. T cell protection against TNFα mediated apoptosis has been found to require IKK [70]. The NFkB family has several roles in the production of IL2, a key cytokine for T cell development. Mice missing p50 do not promote GATA3 expression following T cell stimulation under Th2 differentiation conditions [71]. Moreover, RelB-deficient T cells lack Th1 differentiation, IFNg production, and T-bet expression, most likely due to a failure to upregulate STAT4, which is critical for IFNg to-T-bet signaling.
NFkB is activated by all members of the IL1 protein family (IL1 and IL18), which plays role in development of both Th1 and Th2 responses [72]. In tumor immunity, T cells are arguably the most closely investigated cells. Unlike CD8+ T cells and IFNg-producing Th1 cell, which have significant anti-tumor actions, Foxp3+ regulatory T cells (Treg cells) inhibit immune responses against cancer [73]. NFkB was first identified as a key regulator of T cell homeostasis, which includes proliferation, survival, and cytokine expression [74]. RelA has been identified as a crucial regulator of human CD8+ T cell proliferation [75]. The beginning of FOXP3 expression in immature Treg cells in the thymus is dependent on the activation of RelA and c-Rel via the PKC/CBM/IKK-axis [76,77]. NFkB activation is also essential for mature peripheral Treg cells to maintain homeostasis and identity. This NFkB, master activity in Treg cells has important consequences for cancer immunity. RelA and c-Rel are required for RAR-related orphan receptor (RORgt) expression in developing Th17 cells [78,79]. Overall, evidence suggests that NFkB sits at the crossroads of T cell immunity and tumor tolerance. This provides compelling evidence for using this route as a target in cancer immunotherapy.
Transcription factor NFkB control B cells' ability to survive and mediate their effector roles. c-Rel, RelA, and RelB are necessary for appropriate proliferative responses when the B cell receptor and CD40 are activated [80]. When the BCR is ligated, the activity of NFkB is down-regulated in immature B cells [81]. These cells may be more susceptible to pro-apoptotic signals as a result of lower NFkB activation. The production of pro-survival factors regulated by the conventional and non-canonical NFkB pathways is critical in the last stages of B cell maturation [82]. When NEMO or IKK are lacking, the number of mature B cells is reduced [70]. Similarly, progenitor cells with p50/p52 or RelA/c-Rel double knockouts are unable to develop the B cell transition stage [83]. The activity of NFkB also helps in B cell receptor editing after selection during central tolerance (Figure 2c) [84]. The activities of B lymphocytes in cancer immunity appear to differ depending on the tumor setting. A fraction of B cells with regulatory function (B-regulatory cells) also release inhibitory cytokines such IL10, TGFb, or IL35, which promote tumor growth [85-87]. NFkB is involved in the various aspects of B cell development as well as immune responses [88]. The engagement of the antigen receptor (BCR) can trigger NFkB activation. Despite the huge body of information describing NFkB in non-pathological situations, only a few studies have explicitly investigated NFkB functions in cancer B cells. Human melanoma secretomes have been shown to trigger a large number of NFkB -dependent genes in B cells, including activation markers and co-stimulation molecules like CD30, CD69, or CD137, as well as chemokines like CCL4 and CCL3 [89]. Patients' survival and response to anti-PD1 checkpoint blockade therapy was linked to this enrichment.
NFkB in Inflammation and Tumor Immunity
Inflammation has a role in the development of several cancers. NFkB, as a transcription factor, plays a crucial function in inflammation; it works as a tumor promoter in certain circumstances but also inhibits tumor development in others. In tumor formation, NFkB pathways created and sustained a persistent inflammatory microenvironment [90].
Greten et al. found that mice with IKKb deletion in macrophages had significantly lower chance of developing colitis-associated cancer (CAC). Reduced expression of Bcl-xL, an anti-apoptotic protein targeted by NFkB, causes this [91]. Other studies postulated that the formation of a dominant-negative IkBα "super-repressor" in mice harboring colon or breast cancer cells causes tumor regression and cell death [92]. IKKb-driven NFkB activation in lamina propria macrophages causes CAC development. NFkB stimulates growth factor synthesis, which promotes the proliferation of pre-malignant intestinal epithelial cells (IECs) [91]. Rayet and Gélinas found that hematologic malignancies are associated with prolonged activation of NFκB because of chromosomal rearrangements, overexpression of NFκB subunits, mutations in upstream regulators, or elevated proteosomal activity [93]. Overexpressing the IkBαM super-repressor or deleting RelA or IKK2 in tumor cells in KRAS-induced lung adenocarcinoma reduces tumor growth by inhibiting cell proliferation [94,95]. NFkB plays a role in cancer progression by increasing the production of VEGF, cyclin D1, Twist1, and SNAIL, transcription factors involved in EMT. It also enhances the "Warburg effect" in cancer cells (Figure 3) [10,96-98].

Figure 3. NFkB in inflammation-associated tumor condition. Higher NFkB activity promotes tumor cells to secrete cytokines and chemokines, which attract immune cells towards tumor-site. In immune cells the secreted cytokines activate NFkB to induce transcription of downstream target genes like IL1, IL6, TNFa, VEGF, etc. Immune cells secrete cytokines, which activate NFkB signaling in tumor cells and induce genes like Bcl-xL, Cyclin D1, TWIST, SNAIL, MMP9, VEGF and CXCL2. This leads to tumor cell survival, proliferation, EMT, metastasis, angiogenesis. Developed in BioRender.com.
Blocking NFkB in tumor-associated macrophages (TAMs) can transform the tumor-promoting M2 phenotype to an M1-like phenotype, leading to tumor reduction (Figure 4) [99,100]. Huang et al. found that transfecting IkBαM super-repressor in human prostate cancer cells inhibits NFkB activity, resulting in lower production of VEGF, IL8, and MMP9, reducing tumorigenic and metastatic qualities [101,102]. Kim et al. [103] found that HBV-induced hepatocellular carcinoma (HCC) progression is linked to the activation of NFkB pathways by the oncogenic HBV-X protein via TBK1. In Mdr-deficient mice, prolonged NFkB activation and TNFα generation lead to HCC development. Pikarsky et al. [104] found that transgenic expression of IkBα leads to a failure to develop HCC. According to Park et al. 2010 [105], NFkB plays a significant role in obesity-associated HCC by regulating IL6 and TNFα levels. In DEN-induced HCC, Mx1-Cre-mediated IKK2 deletion in hepatocytes, including Kupffer cells, reduces hepatocarcinogenesis [106]. A rare and aggressive form of breast cancer, inflammatory breast cancer (IBC) occurs by tumor-mediated blockage of the dermal lymphatic pathways. Van Laere et al. [107] found that hyperactivation of EGFR/ErbB2/MAPK is required for activation of NFkB in IBC cells. Van Laere et al. [107] postulated an inverse correlation between NFkB activity and estrogen receptor expression in IBC. Pancreatic ductal adenocarcinoma (PDAC) is characterized by chronic inflammation, which is primarily driven by NFkB. PDAC cells implanted into IRAK4-deficient mice result in a smaller tumor [108]. Chemokine (C-X-C motif) ligand-2 (CXCL2), released by pancreatic stellate cells, activates p50 to inhibit CD8+ T cell infiltration in PDAC [109] (Figure 4). TAMs' NFkB signaling promotes PDAC development. Around 70% of pancreatic tumors exhibit constitutive NFkB activation. Transfection of IkBαM inhibited constitutive NFkB activity and decreased pancreatic carcinogenesis [110].

Figure 4. NFkB in various inflammatory diseases including cancer. NFkB activation has been associated to various illnesses, such as cancer, rheumatoid arthritis, multiple sclerosis, asthma, and inflammatory bowel disease. NFkB plays a key role in cancer progression by regulating gene expression and immune response to malignancies. NFkB triggers inflammatory responses during inflammatory disorders by inducing many inflammatory cytokines and other factors like iNOS COX2 etc. Developed in BioRender.com.
In addition to its involvement in tumor inflammation, NFκB influences the immune responses against tumors. NFkB has been shown to play a pro- and anti-tumorigenic role in a variety of immune cells [111]. T cell canonical NFκB activation is necessary for tumor eradication and increases the quantity of tumor-specific CD8+ T cells that produce IFNγ [112] [113]. It has been demonstrated that NFκB is involved in various NK cell biological processes. The growth of NK cells was inhibited by non-degradable IκBα, which inhibits NFκB. On the other hand, constitutive IKKb activation causes NK cells to become hyperactivated [114]. By generating several molecules like TNFα, IL12, iNOS, COX2, and IL6, NFkB activation can polarize myeloid cells to M1-like macrophages, which oppose carcinogenesis by boosting inflammation, immunostimulation, tissue damage, and cancer cell apoptosis [115]. In dendritic cells, programmed cell death protein-1 (PD1) signaling causes activation of classical NFkB pathway, which in turn inhibits their production of cytokines and expression of co stimulatory molecules [116].
T-regulatory (Treg) cells are immunosuppressive T cells that get amplified in tumor conditions, resulting in decreased anti-tumor immunity [117,118]. According to Oh et al. [77], both the REL and p65 NFkB subunits promote the development and suppressive activity of CD4+CD25+ regulatory T cells. Studies on genetic loss of function in mice demonstrated that signals resulting in NFkB activation and certain NFkB proteins were necessary for Treg cell development and lineage stability [73,119]. With progression of tumor type 2 cytokines like IL4 increases, studies reported role of NFκB in IL4 production [120-123]. Subsequent analysis indicated that the induction of MDSC immunosuppression by TNFα requires the translocation of NFkB p65 and the activation of the NFkB signaling pathway through IκBα degradation [124]. Zhao et al. [125] found that TNFR2 boosted NFkB signaling, leading to improved MDSC survival through upregulation of c-FLIP and suppression of caspase-8 activation. Tu et al. found that IL1β stimulates MDSCs through the IL1RI/ NFkB pathway, leading to immunosuppression and tumor progression [126].
NFkB in Auto-inflammatory Diseases
NFkB is involved in a variety of inflammatory illnesses, including rheumatoid arthritis (RA), inflammatory bowel disease (IBD), multiple sclerosis (MS), atherosclerosis, systemic lupus erythematosus, type-I diabetes, chronic obstructive pulmonary disease, and asthma [127,128]. During inflammatory diseases, NFκB triggers the release of several chemokines in inflammatory tissue, which aid in the recruitment of diverse inflammatory immune cells and ultimately lead to an increase in inflammation (Figure 5). Overexpression of NFkB has been discovered in the inflamed synovium of RA patients. This phenomenon boosts inflammatory cell recruitment and the generation of pro-inflammatory mediators such as IL1, IL6, IL8, and TNFa [129] (Figure 4). The NFkB activation and the release of inflammatory cytokines have been documented in asthma patients also. In bronchial epithelial cells, increased NFkB activity, high production of pro-inflammatory cytokines, chemokines, iNOS, and Cox-2, have been observed [130] (Figure 4). Up-regulation of NFkB has been linked to both IBD and gastritis, caused by Helicobacter pylori. NFkB-mediated inflammation has also been linked to multiple sclerosis and atherosclerosis [131,132].
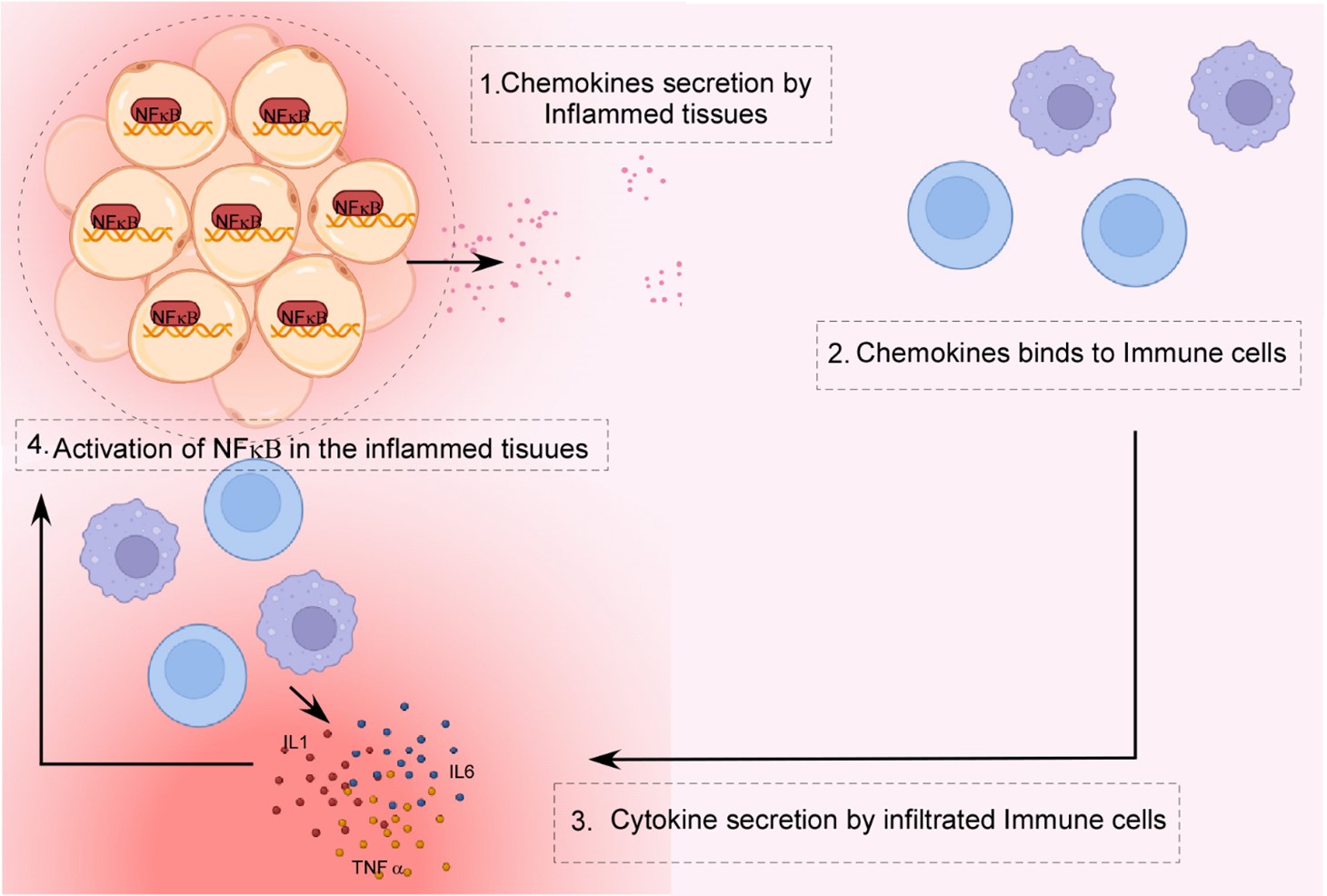
Figure 5. NFkB in autoinflammatory diseases. Overexpression of NFkB in inflamed tissue in autoinflammatory disorders causes the release of a number of chemokines. In response to these chemokines, immune cells are recruited at the site of inflammation. Immune cells further release pro-inflammatory cytokines in inflamed tissues, which are downstream targets of the NFkB signaling. Developed in BioRender.com.
Rheumatoid arthritis, or RA, is an autoimmune and inflammatory disease in which immune cells infiltrate the synovium and cause chronic inflammation, cartilage and bone damage [133]. According to numerous studies, NFkB is a vital inflammatory mediator in RA [134,135]. The pathogenesis involving monocytes, macrophages, T cells, B cells, and synovial fibroblasts requires the deregulation of NFkB [136]. In macrophages, NFkB stimulates the secretion of various pro-inflammatory cytokines like TNFα, IL1, and IL6 [137]. Many of these cytokines have the ability to stimulate NFkB in innate immune cells leading to more inflammatory cytokines and chemokines generation. It resulted in the recruitment of inflammatory immune cells and the spread of inflammation. The abnormality in RANK ligand-induced differentiation of monocytes and macrophages by both canonical and non-canonical NFkB pathways develop an inflammatory bone loss in RA patients [138]. It is a well-known fact that Th17 cells are critical for the etiology of RA, and NFkB promotes Th17 development by generating inflammatory cytokines such as IL1, IL6, and IL23 [79,139,140]. Furthermore, NFkB activation assists in abnormal self-reactive B cells to survive and stimulate autoantibody secretion, which contributes to the pathogenesis of RA in patients [141].
Several common chronic inflammatory bowel disorders, such as Crohn’s disease and ulcerative colitis, are developed from the unwanted inflammatory responses to intestinal microbes [142]. Multiple mucosal immune system cell types are involved in IBD development [143]. It is widely known that NFkB plays a role in the etiology of IBD. Constant NFkB expression has been detected in the inflamed colonic mucosa of IBD patients [144,145]. Furthermore, polymorphisms or mutations in the genes of NFkB-regulating factors, such as NOD2, IL12, IL23, IkBα-like protein molecules, p105, p50, are linked to human IBD condition [146,147]. These genetic alterations alter the stability and functionality of the protein products and suppress the expression of the NFkB1 gene. Mice with a knockout mutation in the NFkB1 gene produce less p105, leading to more IBD-like intestinal inflammation [148]. These findings support the role of NFkB in pro-inflammatory cytokines secretion in innate immune cells and the development of Th1 and Th17 subsets of inflammatory T cells. In mice model, experimental deletion of NEMO, IKKb or both IKKa and IKKb in intestinal epithelial cells results in the development of chronic intestinal inflammation [149,150]. Therefore, abnormal stimulation of NFkB or any genetic modification might lead to IBD.
A central nervous system (CNS) autoimmune disease is multiple sclerosis (MS). It is an inflammatory condition caused by CNS-specific CD4+ T cells, mostly Th1 and Th17 cells, acting in a pathogenic manner [151]. The NFkB signaling pathway has been linked to MS patients in genome-wide association studies. RelA, NIK, Bcl10, and MALT1 are only a few of the NFkB-related variables that have been identified as susceptibility candidates [152]. In the pathophysiology of experimental autoimmune encephalomyelitis (EAE), an established animal model of MS, both the canonical and non-canonical NFkB pathways play important roles. T cell-specific deletion or inhibition of IKKb reduces mice refractory to EAE initiation [153]. Correspondingly, a genetic anomaly in IKK upstream signaling factors, i.e. CARMA1 and MALT1 of the TCR pathway enhance EAE stimulation [154]. RORgt, Th17-lineage transcription factor, and the classical NFkB members RelA and c-Rel increase its expression [155,156]. In Th17 cells, p52 regulates the expression of the inflammatory cytokine GM-CSF in combination with c-Rel, the non-canonical NFkB component [156]. In myeloid cells, the deletion of IkBα via the LysM-Cre system causes constitutive activation of NFkB, causing myelin oligodendrocyte glycoprotein-induced EAE to cause more severe CNS inflammation [157]. Instead, myeloid cell-specific deletion of IKKb reduces EAE stimulation, which is associated with a reduced generation of inflammatory Th1 and Th17 cells [158]. Meanwhile, conditional ablation of NEMO or IKKb prevents EAE induction to some extent [159]. Furthermore, transgenic production of a degradation-resistant variant of IkBα (IkB-dn) suppresses inflammatory cytokine expression in astrocytes and reduces the pathogenic severity of EAE [160,161].
Atherosclerosis is an inflammatory condition of the artery wall that progresses over time. It occurred as a result of immune cells and low-density lipoprotein (LDL) building up in the sub-endothelium region. Endothelial cells, monocytes, and T cells are among the cell types involved in the pathophysiology of atherosclerosis [162]. The initiation of atherosclerosis is believed to be mediated by endothelial cell activation, which releases chemotactic proteins and cell adhesion molecules that allow blood monocytes to aggregate inside the artery intima. The monocytes there transform into macrophages, which then suck up LDL molecules. They eventually transformed into lipid-laden foam cells, which play a role in the formation of atherosclerotic plaques. NFkB regulates the expression of several genes implicated in the pathophysiology of atherosclerosis [163]. NFkB stimulates the production of pro-inflammatory cytokines, chemotactic factors, and adhesion molecules in vascular endothelial cells, which aids monocyte recruitment and disease progression [164,165]. It has been demonstrated that chemokine expression and monocyte accumulation can be inhibited in endothelial cells by conditional deletion of NEMO or transgenic expression of a degradation-resistant IκBα, which lessens the severity of atherosclerosis. [166]. In myeloid cells, NFkB was linked to increased inflammatory gene expression and macrophage transformation into foam cells [163]. Interestingly, deletion of IKKb in myeloid cells reduces atherosclerotic lesion areas in LDL receptor-deficient animals in numerous studies, whereas deletion of IKKb in myeloid cells enlarges atherosclerotic lesion diameters in the same mouse model in another study [167,168]. The explanation for the discrepancy in results is still unknown, though it is presumed that the mismatch in the two experimental methodologies is to blame.
Conclusion
As we understand more about our immune system, we're finding how important it is for pathogen avoidance, pathogen destruction, and intricately regulating a homeostasis to keep the organism healthy, despite the fact that many inflammatory disorders are emerging. In search of the source of these devastating diseases, intense research works are showing how a single NFkB family is connected with almost all immune responses particularly in inflammation. Deregulated NFkB causes malfunctioning of proper immune response generation. Thus, NFκB signaling targeting may present a promising therapeutic strategy for anti-inflammatory treatments. Numerous inhibitors have been created to block various up or downstream steps in NFkB signaling like selective IKK inhibitors[169], some well-known drugs such as aspirin and salicylate can inhibit IKK [170], Proteasome inhibitors, one example is Velcade (also called Bortezomib and PS-341) and lactacystin, which stop degradation of IkBα in the proteasome [171], inhibitors that blocking nuclear translocation of NFkB subunits, like tacrolimus (FK-506) etc. [172]. Despite of tremendous achievements in the designing of different blockers to inhibit NFkB as anti-inflammatory drugs, scarcity of available clinical medicine for NFkB blockers is still a daunting situation. As total inhibition of NFkB may cause severe side effects because of many normal cells functioning like cell survival, apoptosis depends on NFkB signaling, therefore gaining deep knowledge underlying the mechanism of pathological or deregulated activation or functioning of NFkB signaling for the development of more targeted and efficient therapies for treating inflammatory illnesses is quite alarming and urgent requirement looking at the present scenario.
Ethical Approval and Consent to Participate
Not applicable.
Competing Interests
The authors declare that they have no competing interests.
Authors' Contributions
SM and SD did the background literature study and prepared the initial draft; SP developed illustrations; TS, SP, SC, UB, SM and SD contributed to literature study and extending the draft and arranging the references. TD made the final editing of the draft and GS conceptualized supervised the entire project.
Acknowledgment
GS [HRD/IES/2023(01)] and TD [74/1/2020-Pers] are ICMR Emeritus Scientists. The study was funded by Indian Council for Medical Research, Government of India. We acknowledge BioRender platform (https://www.biorender.com) for creation of the illustrations.
References
2. Zheng D, Liwinski T, Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020 Jun;30(6):492-506.
3. Wu Y, Zheng Z, Jiang Y, Chess L, Jiang H. The specificity of T cell regulation that enables self-nonself discrimination in the periphery. Proc Natl Acad Sci U S A. 2009 Jan 13;106(2):534-9.
4. Ribas A. Adaptive Immune Resistance: How Cancer Protects from Immune Attack. Cancer Discov. 2015 Sep;5(9):915-9.
5. Zhu Y, Yao S, Chen L. Cell surface signaling molecules in the control of immune responses: a tide model. Immunity. 2011 Apr 22;34(4):466-78.
6. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009 Apr;22(2):240-73.
7. Smale ST. Transcriptional regulation in the immune system: a status report. Trends Immunol. 2014 May;35(5):190-4.
8. Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009 Oct;1(4):a000034.
9. Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006 Oct 30;25(51):6758-80.
10. Taniguchi K, Karin M. NF-κB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018 May;18(5):309-24.
11. Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693-733.
12. Sun SC, Liu ZG. A special issue on NF-κB signaling and function. Cell Res. 2011;21:1-2.
13. Sun SC. Non-canonical NF-κB signaling pathway. Cell Res. 2011 Jan;21(1):71-85.
14. Zhang H, Sun SC. NF-κB in inflammation and renal diseases. Cell Biosci. 2015 Nov 16;5:63.
15. Karin M, Delhase M. The I kappa B kinase (IKK) and NF-kappa B: key elements of proinflammatory signalling. Semin Immunol. 2000 Feb;12(1):85-98.
16. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008 Feb 8;132(3):344-62.
17. Cildir G, Low KC, Tergaonkar V. Noncanonical NF-κB Signaling in Health and Disease. Trends Mol Med. 2016 May;22(5):414-29.
18. Senftleben U, Cao Y, Xiao G, Greten FR, Krähn G, Bonizzi G, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001 Aug 24;293(5534):1495-9.
19. Eck SL, Perkins ND, Carr DP, Nabel GJ. Inhibition of phorbol ester-induced cellular adhesion by competitive binding of NF-kappa B in vivo. Mol Cell Biol. 1993 Oct;13(10):6530-6.
20. Vincenti MP, Brinckerhoff CE. Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res. 2002;4(3):157-64.
21. Lawrence T, Gilroy DW, Colville-Nash PR, Willoughby DA. Possible new role for NF-kappaB in the resolution of inflammation. Nat Med. 2001 Dec;7(12):1291-7.
22. Silverman N, Maniatis T. NF-kappaB signaling pathways in mammalian and insect innate immunity. Genes Dev. 2001 Sep 15;15(18):2321-42.
23. Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012 Mar 1;4(3):a006049.
24. Liang Y, Zhou Y, Shen P. NF-kappaB and its regulation on the immune system. Cell Mol Immunol. 2004 Oct;1(5):343-50.
25. Hatada EN, Krappmann D, Scheidereit C. NF-kappaB and the innate immune response. Curr Opin Immunol. 2000 Feb;12(1):52-8.
26. Caamaño J, Hunter CA. NF-kappaB family of transcription factors: central regulators of innate and adaptive immune functions. Clin Microbiol Rev. 2002 Jul;15(3):414-29.
27. Xia YF, Liu LP, Zhong CP, Geng JG. NF-kappaB activation for constitutive expression of VCAM-1 and ICAM-1 on B lymphocytes and plasma cells. Biochem Biophys Res Commun. 2001 Dec 14;289(4):851-6.
28. Park YC, Rimbach G, Saliou C, Valacchi G, Packer L. Activity of monomeric, dimeric, and trimeric flavonoids on NO production, TNF-alpha secretion, and NF-kappaB-dependent gene expression in RAW 264.7 macrophages. FEBS Lett. 2000 Jan 14;465(2-3):93-7.
29. Diamond G, Kaiser V, Rhodes J, Russell JP, Bevins CL. Transcriptional regulation of beta-defensin gene expression in tracheal epithelial cells. Infect Immun. 2000 Jan;68(1):113-9.
30. Pennington KN, Taylor JA, Bren GD, Paya CV. IkappaB kinase-dependent chronic activation of NF-kappaB is necessary for p21(WAF1/Cip1) inhibition of differentiation-induced apoptosis of monocytes. Mol Cell Biol. 2001 Mar;21(6):1930-41.
31. Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature. 2005 Apr 28;434(7037):1138-43.
32. Li Q, Lu Q, Bottero V, Estepa G, Morrison L, Mercurio F, et al. Enhanced NF-kappaB activation and cellular function in macrophages lacking IkappaB kinase 1 (IKK1). Proc Natl Acad Sci U S A. 2005 Aug 30;102(35):12425-30.
33. Wang N, Liang H, Zen K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol. 2014 Nov 28;5:614.
34. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012 Mar;122(3):787-95.
35. Mosser DM. The many faces of macrophage activation. J Leukoc Biol. 2003 Feb;73(2):209-12.
36. Yu M, Zhou H, Zhao J, Xiao N, Roychowdhury S, Schmitt D, et al. MyD88-dependent interplay between myeloid and endothelial cells in the initiation and progression of obesity-associated inflammatory diseases. J Exp Med. 2014 May 5;211(5):887-907.
37. Sanjabi S, Hoffmann A, Liou HC, Baltimore D, Smale ST. Selective requirement for c-Rel during IL-12 P40 gene induction in macrophages. Proc Natl Acad Sci U S A. 2000 Nov 7;97(23):12705-10.
38. Courtine E, Cagnard N, Mazzolini J, Antona M, Pène F, Fitting C, et al. Combined loss of cRel/p50 subunits of NF-κB leads to impaired innate host response in sepsis. Innate Immun. 2012 Oct;18(5):753-63.
39. Huang Y, Krein PM, Muruve DA, Winston BW. Complement factor B gene regulation: synergistic effects of TNF-alpha and IFN-gamma in macrophages. J Immunol. 2002 Sep 1;169(5):2627-35.
40. Shih VF, Davis-Turak J, Macal M, Huang JQ, Ponomarenko J, Kearns JD, et al. Control of RelB during dendritic cell activation integrates canonical and noncanonical NF-κB pathways. Nat Immunol. 2012 Dec;13(12):1162-70.
41. Li M, Zhang X, Zheng X, Lian D, Zhang ZX, Ge W, et al. Immune modulation and tolerance induction by RelB-silenced dendritic cells through RNA interference. J Immunol. 2007 May 1;178(9):5480-7.
42. Platzer B, Jörgl A, Taschner S, Höcher B, Strobl H. RelB regulates human dendritic cell subset development by promoting monocyte intermediates. Blood. 2004 Dec 1;104(12):3655-63.
43. Boffa DJ, Feng B, Sharma V, Dematteo R, Miller G, Suthanthiran M, et al. Selective loss of c-Rel compromises dendritic cell activation of T lymphocytes. Cell Immunol. 2003 Apr;222(2):105-15.
44. Ouaaz F, Arron J, Zheng Y, Choi Y, Beg AA. Dendritic cell development and survival require distinct NF-kappaB subunits. Immunity. 2002 Feb;16(2):257-70.
45. Kriehuber E, Bauer W, Charbonnier AS, Winter D, Amatschek S, Tamandl D, et al. Balance between NF-kappaB and JNK/AP-1 activity controls dendritic cell life and death. Blood. 2005 Jul 1;106(1):175-83.
46. Kobayashi T, Walsh PT, Walsh MC, Speirs KM, Chiffoleau E, King CG, et al. TRAF6 is a critical factor for dendritic cell maturation and development. Immunity. 2003 Sep;19(3):353-63.
47. Yoshimura S, Bondeson J, Foxwell BM, Brennan FM, Feldmann M. Effective antigen presentation by dendritic cells is NF-kappaB dependent: coordinate regulation of MHC, co-stimulatory molecules and cytokines. Int Immunol. 2001 May;13(5):675-83.
48. Li M, Carpio DF, Zheng Y, Bruzzo P, Singh V, Ouaaz F, et al. An essential role of the NF-kappa B/Toll-like receptor pathway in induction of inflammatory and tissue-repair gene expression by necrotic cells. J Immunol. 2001 Jun 15;166(12):7128-35.
49. McDonald PP, Bald A, Cassatella MA. Activation of the NF-kappaB pathway by inflammatory stimuli in human neutrophils. Blood. 1997 May 1;89(9):3421-33.
50. Choi M, Rolle S, Wellner M, Cardoso MC, Scheidereit C, Luft FC, et al. Inhibition of NF-kappaB by a TAT-NEMO-binding domain peptide accelerates constitutive apoptosis and abrogates LPS-delayed neutrophil apoptosis. Blood. 2003 Sep 15;102(6):2259-67.
51. François S, El Benna J, Dang PM, Pedruzzi E, Gougerot-Pocidalo MA, Elbim C. Inhibition of neutrophil apoptosis by TLR agonists in whole blood: involvement of the phosphoinositide 3-kinase/Akt and NF-kappaB signaling pathways, leading to increased levels of Mcl-1, A1, and phosphorylated Bad. J Immunol. 2005 Mar 15;174(6):3633-42.
52. McDonald PP, Cassatella MA. Activation of transcription factor NF-κB by phagocytic stimuli in human neutrophils. FEBS Letters. 1997 August 4;412(3):583-6.
53. Castro-Alcaraz S, Miskolci V, Kalasapudi B, Davidson D, Vancurova I. NF-kappa B regulation in human neutrophils by nuclear I kappa B alpha: correlation to apoptosis. J Immunol. 2002 Oct 1;169(7):3947-53.
54. Siebenlist U, Brown K, Claudio E. Control of lymphocyte development by nuclear factor-kappaB. Nat Rev Immunol. 2005 Jun;5(6):435-45.
55. Grumont RJ, Rourke IJ, O'Reilly LA, Strasser A, Miyake K, Sha W, et al. B lymphocytes differentially use the Rel and nuclear factor kappaB1 (NF-kappaB1) transcription factors to regulate cell cycle progression and apoptosis in quiescent and mitogen-activated cells. J Exp Med. 1998 Mar 2;187(5):663-74.
56. Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat Immunol. 2002 Oct;3(10):958-65.
57. Prendes M, Zheng Y, Beg AA. Regulation of developing B cell survival by RelA-containing NF-kappa B complexes. J Immunol. 2003 Oct 15;171(8):3963-9.
58. Swallow MM, Wallin JJ, Sha WC. B7h, a novel costimulatory homolog of B7.1 and B7.2, is induced by TNFalpha. Immunity. 1999 Oct;11(4):423-32.
59. Hettmann T, DiDonato J, Karin M, Leiden JM. An essential role for nuclear factor kappaB in promoting double positive thymocyte apoptosis. J Exp Med. 1999 Jan 4;189(1):145-58.
60. Mora AL, Stanley S, Armistead W, Chan AC, Boothby M. Inefficient ZAP-70 phosphorylation and decreased thymic selection in vivo result from inhibition of NF-kappaB/Rel. J Immunol. 2001 Nov 15;167(10):5628-35.
61. French LE, Hahne M, Viard I, Radlgruber G, Zanone R, Becker K, et al. Fas and Fas ligand in embryos and adult mice: ligand expression in several immune-privileged tissues and coexpression in adult tissues characterized by apoptotic cell turnover. J Cell Biol. 1996 Apr;133(2):335-43.
62. Ouaaz F, Li M, Beg AA. A critical role for the RelA subunit of nuclear factor kappaB in regulation of multiple immune-response genes and in Fas-induced cell death. J Exp Med. 1999 Mar 15;189(6):999-1004.
63. Rescigno M, Martino M, Sutherland CL, Gold MR, Ricciardi-Castagnoli P. Dendritic cell survival and maturation are regulated by different signaling pathways. J Exp Med. 1998 Dec 7;188(11):2175-80.
64. Sun Z, Arendt CW, Ellmeier W, Schaeffer EM, Sunshine MJ, Gandhi L, et al. PKC-theta is required for TCR-induced NF-kappaB activation in mature but not immature T lymphocytes. Nature. 2000 Mar 23;404(6776):402-7.
65. Murphy TL, Cleveland MG, Kulesza P, Magram J, Murphy KM. Regulation of interleukin 12 p40 expression through an NF-kappa B half-site. Mol Cell Biol. 1995 Oct;15(10):5258-67.
66. Ivanov VN, Nikolić-Zugić J. Transcription factor activation during signal-induced apoptosis of immature CD4(+)CD8(+) thymocytes. A protective role of c-Fos. J Biol Chem. 1997 Mar 28;272(13):8558-66.
67. Jeremias I, Kupatt C, Baumann B, Herr I, Wirth T, Debatin KM. Inhibition of nuclear factor kappaB activation attenuates apoptosis resistance in lymphoid cells. Blood. 1998 Jun 15;91(12):4624-31.
68. Bhattacharyya S, Mandal D, Sen GS, Pal S, Banerjee S, Lahiry L, et al. Tumor-induced oxidative stress perturbs nuclear factor-kappaB activity-augmenting tumor necrosis factor-alpha-mediated Bdeath: protection by curcumin. Cancer Res. 2007 Jan 1;67(1):362-70.
69. Sa G, Das T, Moon C, Hilston CM, Rayman PA, Rini BI, et al. GD3, an overexpressed tumor-derived ganglioside, mediates the apoptosis of activated but not resting T cells. Cancer Res. 2009 Apr 1;69(7):3095-104.
70. Senftleben U, Li ZW, Baud V, Karin M. IKKbeta is essential for protecting T cells from TNFalpha-induced apoptosis. Immunity. 2001 Mar;14(3):217-30.
71. Das J, Chen CH, Yang L, Cohn L, Ray P, Ray A. A critical role for NF-kappa B in GATA3 expression and TH2 differentiation in allergic airway inflammation. Nat Immunol. 2001 Jan;2(1):45-50.
72. Huber M, Beuscher HU, Rohwer P, Kurrle R, Röllinghoff M, Lohoff M. Costimulation via TCR and IL-1 receptor reveals a novel IL-1alpha-mediated autocrine pathway of Th2 cell proliferation. J Immunol. 1998 May 1;160(9):4242-7.
73. Sarkar T, Dhar S, Chakraborty D, Pati S, Bose S, Panda AK, et al. FOXP3/HAT1 Axis Controls Treg Infiltration in the Tumor Microenvironment by Inducing CCR4 Expression in Breast Cancer. Front Immunol. 2022 Feb 9;13:740588.
74. Ronin E, Lubrano di Ricco M, Vallion R, Divoux J, Kwon HK, Grégoire S, et al. The NF-κB RelA Transcription Factor Is Critical for Regulatory T Cell Activation and Stability. Front Immunol. 2019 Oct 30;10:2487.
75. Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, et al. Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell. 2018 Dec 13;175(7):1958-71.e15.
76. Isomura I, Palmer S, Grumont RJ, Bunting K, Hoyne G, Wilkinson N, et al. c-Rel is required for the development of thymic Foxp3+ CD4 regulatory T cells. J Exp Med. 2009 Dec 21;206(13):3001-14.
77. Oh H, Grinberg-Bleyer Y, Liao W, Maloney D, Wang P, Wu Z, et al. An NF-κB Transcription-Factor-Dependent Lineage-Specific Transcriptional Program Promotes Regulatory T Cell Identity and Function. Immunity. 2017 Sep 19;47(3):450-65.e5.
78. Chen G, Hardy K, Pagler E, Ma L, Lee S, Gerondakis S, et al. The NF-κB transcription factor c-Rel is required for Th17 effector cell development in experimental autoimmune encephalomyelitis. J Immunol. 2011 Nov 1;187(9):4483-91.
79. Roy D, Bose S, Pati S, Guin A, Banerjee K, Saha S, et al. GFI1/HDAC1-axis differentially regulates immunosuppressive CD73 in human tumor-associated FOXP3+ Th17 and inflammation-linked Th17 cells. Eur J Immunol. 2021 May;51(5):1206-1217.
80. Snapper CM, Rosas FR, Zelazowski P, Moorman MA, Kehry MR, Bravo R, et al. B cells lacking RelB are defective in proliferative responses, but undergo normal B cell maturation to Ig secretion and Ig class switching. J Exp Med. 1996 Oct 1;184(4):1537-41.
81. Wu M, Lee H, Bellas RE, Schauer SL, Arsura M, Katz D, et al. Inhibition of NF-kappaB/Rel induces apoptosis of murine B cells. EMBO J. 1996 Sep 2;15(17):4682-90.
82. Grossmann M, O'Reilly LA, Gugasyan R, Strasser A, Adams JM, Gerondakis S. The anti-apoptotic activities of Rel and RelA required during B-cell maturation involve the regulation of Bcl-2 expression. EMBO J. 2000 Dec 1;19(23):6351-60.
83. Franzoso G, Carlson L, Xing L, Poljak L, Shores EW, Brown KD, et al. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 1997 Dec 15;11(24):3482-96.
84. Cadera EJ, Wan F, Amin RH, Nolla H, Lenardo MJ, Schlissel MS. NF-kappaB activity marks cells engaged in receptor editing. J Exp Med. 2009 Aug 3;206(8):1803-16.
85. Inoue S, Leitner WW, Golding B, Scott D. Inhibitory effects of B cells on antitumor immunity. Cancer Res. 2006 Aug 1;66(15):7741-7.
86. Pati S, Chowdhury A, Mukherjee S, Guin A, Mukherjee S, Sa G. Regulatory lymphocytes: the dice that resolve the tumor endgame. Applied Cancer Research. 2020 Dec;40:7.
87. Pati S, Mukherjee S, Dutta S, Guin A, Roy D, Bose S, et al. Tumor-Associated CD19+CD39- B Regulatory Cells Deregulate Class-Switch Recombination to Suppress Antibody Responses. Cancer Immunol Res. 2023 Mar 1;11(3):364-80.
88. Sasaki Y, Iwai K. Roles of the NF-κB Pathway in B-Lymphocyte Biology. Curr Top Microbiol Immunol. 2016;393:177-209.
89. Griss J, Bauer W, Wagner C, Simon M, Chen M, Grabmeier-Pfistershammer K, et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat Commun. 2019 Sep 13;10(1):4186.
90. Ditsworth D, Zong WX. NF-kappaB: key mediator of inflammation-associated cancer. Cancer Biol Ther. 2004 Dec;3(12):1214-6.
91. Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004 Aug 6;118(3):285-96.
92. Harmey JH, Bucana CD, Lu W, Byrne AM, McDonnell S, Lynch C, et al. Lipopolysaccharide-induced metastatic growth is associated with increased angiogenesis, vascular permeability and tumor cell invasion. Int J Cancer. 2002 Oct 10;101(5):415-22.
93. Rayet B, Gélinas C. Aberrant rel/nfkb genes and activity in human cancer. Oncogene. 1999;18:6938-47.
94. Xia Y, Yeddula N, Leblanc M, Ke E, Zhang Y, Oldfield E, et al. Reduced cell proliferation by IKK2 depletion in a mouse lung-cancer model. Nat Cell Biol. 2012;4:257-65.
95. Bassères DS, Ebbs A, Levantini E, Baldwin AS. Requirement of the NF-kappaB subunit p65/RelA for K-Ras-induced lung tumorigenesis. Cancer Res. 2010;70:3537-46.
96. Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996 Sep;16(9):4604-13.
97. Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, et al. Epithelial-mesenchymal transition induced by TNF-α requires NF-κB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012 Mar 1;72(5):1290-300.
98. Mauro C, Leow SC, Anso E, Rocha S, Thotakura AK, Tornatore L, et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat Cell Biol. 2011 Aug 28;13(10):1272-9.
99. Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, et al. "Re-educating" tumor-associated macrophages by targeting NF-kappaB. J Exp Med. 2008 Jun 9;205(6):1261-8.
100. Basak U, Sarkar T, Mukherjee S, Chakraborty S, Dutta A, Dutta S, et al. Tumor-associated macrophages: an effective player of the tumor microenvironment. Front Immunol. 2023 Nov 16;14:1295257.
101. Huang S, Pettaway CA, Uehara H, Bucana CD, Fidler IJ. Blockade of NF-kappaB activity in human prostate cancer cells is associated with suppression of angiogenesis, invasion, and metastasis. Oncogene. 2001 Jul 12;20(31):4188-97.
102. Kajal K, Bose S, Panda AK, Chakraborty D, Chakraborty S, Pati S, et al. Transcriptional regulation of VEGFA expression in T-regulatory cells from breast cancer patients. Cancer Immunol Immunother. 2021 Jul;70(7):1877-1891.
103. Kim HR, Lee SH, Jung G. The hepatitis B viral X protein activates NF-kappaB signaling pathway through the up-regulation of TBK1. FEBS Lett. 2010 Feb 5;584(3):525-30.
104. Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004 Sep 23;431(7007):461-6.
105. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010 Jan 22;140(2):197-208.
106. Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005 Jul 1;121(7):977-90.
107. Van Laere SJ, Van der Auwera I, Van den Eynden GG, van Dam P, Van Marck EA, Vermeulen PB, et al. NF-kappaB activation in inflammatory breast cancer is associated with oestrogen receptor downregulation, secondary to EGFR and/or ErbB2 overexpression and MAPK hyperactivation. Br J Cancer. 2007 Sep 3;97(5):659-69.
108. Zhang D, Li L, Jiang H, Li Q, Wang-Gillam A, Yu J, et al. Tumor-Stroma IL1β-IRAK4 Feedforward Circuitry Drives Tumor Fibrosis, Chemoresistance, and Poor Prognosis in Pancreatic Cancer. Cancer Res. 2018 Apr 1;78(7):1700-12.
109. Garg B, Giri B, Modi S, Sethi V, Castro I, Umland O, et al. NFκB in Pancreatic Stellate Cells Reduces Infiltration of Tumors by Cytotoxic T Cells and Killing of Cancer Cells, via Up-regulation of CXCL12. Gastroenterology. 2018;155:880-91.e8.
110. Fujioka S, Sclabas GM, Schmidt C, Frederick WA, Dong QG, Abbruzzese JL, et al. Function of nuclear factor kappaB in pancreatic cancer metastasis. Clin Cancer Res. 2003 Jan;9(1):346-54.
111. Paul S, Sa G. Curcumin as an Adjuvant to Cancer Immunotherapy. Front Oncol. 2021 Aug 16;11:675923.
112. Evaristo C, Spranger S, Barnes SE, Miller ML, Molinero LL, Locke FL, et al. Cutting Edge: Engineering Active IKKβ in T Cells Drives Tumor Rejection. J Immunol. 2016 Apr 1;196(7):2933-8.
113. Chakraborty S, Bhattacharjee P, Panda AK, Kajal K, Bose S, Sa G. Providence of the CD25+ KIR+ CD127- FOXP3- CD8+ T-cell subset determines the dynamics of tumor immune surveillance. Immunol Cell Biol. 2018 Nov;96(10):1035-1048. Chakraborty S, Bhattacharjee P, Panda AK, Kajal K, Bose S, Sa G. Providence of the CD25+ KIR+ CD127- FOXP3- CD8+ T-cell subset determines the dynamics of tumor immune surveillance. Immunol Cell Biol. 2018 Nov;96(10):1035-1048.
114. Lalle G, Twardowski J, Grinberg-Bleyer Y. NF-κB in Cancer Immunity: Friend or Foe? Cells. 2021 Feb 9;10(2):355.
115. Cornice J, Verzella D, Arboretto P, Vecchiotti D, Capece D, Zazzeroni F, et al. NF-κB: Governing Macrophages in Cancer. Genes (Basel). 2024 Jan 31;15(2):197.
116. Karyampudi L, Lamichhane P, Krempski J, Kalli KR, Behrens MD, Vargas DM, et al. PD-1 Blunts the Function of Ovarian Tumor-Infiltrating Dendritic Cells by Inactivating NF-κB. Cancer Res. 2016 Jan 15;76(2):239-50.
117. Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019 Jul;110(7):2080-9.
118. Mukherjee S, Chakraborty S, Basak U, Pati S, Dutta A, Dutta S, et al. Breast cancer stem cells generate immune-suppressive T regulatory cells by secreting TGFβ to evade immune-elimination. Discov Oncol. 2023 Dec 1;14(1):220.
119. Hövelmeyer N, Schmidt-Supprian M, Ohnmacht C. NF-κB in control of regulatory T cell development, identity, and function. J Mol Med (Berl). 2022 Jul;100(7):985-95.
120. Guenova E, Watanabe R, Teague JE, Desimone JA, Jiang Y, Dowlatshahi M, et al. TH2 cytokines from malignant cells suppress TH1 responses and enforce a global TH2 bias in leukemic cutaneous T-cell lymphoma. Clin Cancer Res. 2013 Jul 15;19(14):3755-63.
121. Bhattacharyya S, Md Sakib Hossain D, Mohanty S, Sankar Sen G, Chattopadhyay S, Banerjee S, et al. Curcumin reverses T cell-mediated adaptive immune dysfunctions in tumor-bearing hosts. Cell Mol Immunol. 2010 Jul;7(4):306-15.
122. Bose S, Panda AK, Mukherjee S, Sa G. Curcumin and tumor immune-editing: resurrecting the immune system. Cell Div. 2015 Oct 12;10:6.
123. Li-Weber M, Giaisi M, Baumann S, Pálfi K, Krammer PH. NF-kappa B synergizes with NF-AT and NF-IL6 in activation of the IL-4 gene in T cells. Eur J Immunol. 2004 Apr;34(4):1111-8.
124. Hu X, Li B, Li X, Zhao X, Wan L, Lin G, et al. Transmembrane TNF-α promotes suppressive activities of myeloid-derived suppressor cells via TNFR2. J Immunol. 2014 Feb 1;192(3):1320-31.
125. Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J, et al. TNF signaling drives myeloid-derived suppressor cell accumulation. J Clin Invest. 2012 Nov;122(11):4094-104.
126. Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008 Nov 4;14(5):408-19.
127. Pai S, Thomas R. Immune deficiency or hyperactivity-Nf-kappab illuminates autoimmunity. J Autoimmun. 2008 Nov;31(3):245-51.
128. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023.
129. Han Z, Boyle DL, Manning AM, Firestein GS. AP-1 and NF-kappaB regulation in rheumatoid arthritis and murine collagen-induced arthritis. Autoimmunity. 1998;28(4):197-208.
130. Hart LA, Krishnan VL, Adcock IM, Barnes PJ, Chung KF. Activation and localization of transcription factor, nuclear factor-kappaB, in asthma. Am J Respir Crit Care Med. 1998 Nov;158(5 Pt 1):1585-92.
131. Mattson MP, Camandola S. NF-kappaB in neuronal plasticity and neurodegenerative disorders. J Clin Invest. 2001 Feb;107(3):247-54.
132. Collins T, Cybulsky MI. NF-kappaB: pivotal mediator or innocent bystander in atherogenesis? J Clin Invest. 2001 Feb;107(3):255-64.
133. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. 2011 Dec 8;365(23):2205-19.
134. Marok R, Winyard PG, Coumbe A, Kus ML, Gaffney K, Blades S, et al. Activation of the transcription factor nuclear factor-kappaB in human inflamed synovial tissue. Arthritis Rheum. 1996 Apr;39(4):583-91.
135. Gilston V, Jones HW, Soo CC, Coumbe A, Blades S, Kaltschmidt C, et al. NF-kappa B activation in human knee-joint synovial tissue during the early stage of joint inflammation. Biochem Soc Trans. 1997 Aug;25(3):518S.
136. Feldmann M, Maini RN. Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol. 2001;19:163-96.
137. Davignon JL, Hayder M, Baron M, Boyer JF, Constantin A, Apparailly F, et al. Targeting monocytes/macrophages in the treatment of rheumatoid arthritis. Rheumatology (Oxford). 2013 Apr;52(4):590-8.
138. Novack DV. Role of NF-κB in the skeleton. Cell Res. 2011 Jan;21(1):169-82.
139. Mellado M, Martínez-Muñoz L, Cascio G, Lucas P, Pablos JL, Rodríguez-Frade JM. T Cell Migration in Rheumatoid Arthritis. Front Immunol. 2015 Jul 27;6:384.
140. Teng MW, Bowman EP, McElwee JJ, Smyth MJ, Casanova JL, Cooper AM, et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat Med. 2015 Jul;21(7):719-29.
141. Wei F, Chang Y, Wei W. The role of BAFF in the progression of rheumatoid arthritis. Cytokine. 2015 Dec;76(2):537-44.
142. Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009 Nov 19;361(21):2066-78.
143. Wallace KL, Zheng LB, Kanazawa Y, Shih DQ. Immunopathology of inflammatory bowel disease. World J Gastroenterol. 2014 Jan 7;20(1):6-21.
144. Rogler G, Brand K, Vogl D, Page S, Hofmeister R, Andus T, et al. Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology. 1998 Aug;115(2):357-69.
145. Schreiber S, Nikolaus S, Hampe J. Activation of nuclear factor kappa B inflammatory bowel disease. Gut. 1998 Apr;42(4):477-84.
146. Karban AS, Okazaki T, Panhuysen CI, Gallegos T, Potter JJ, Bailey-Wilson JE, et al. Functional annotation of a novel NFKB1 promoter polymorphism that increases risk for ulcerative colitis. Hum Mol Genet. 2004 Jan 1;13(1):35-45.
147. Kaustio M, Haapaniemi E, Göös H, Hautala T, Park G, Syrjänen J, et al. Damaging heterozygous mutations in NFKB1 lead to diverse immunologic phenotypes. J Allergy Clin Immunol. 2017 Sep;140(3):782-796.
148. Chang M, Lee AJ, Fitzpatrick L, Zhang M, Sun SC. NF-kappa B1 p105 regulates T cell homeostasis and prevents chronic inflammation. J Immunol. 2009 Mar 1;182(5):3131-8.
149. Nenci A, Becker C, Wullaert A, Gareus R, van Loo G, Danese S, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007 Mar 29;446(7135):557-61.
150. Zaph C, Troy AE, Taylor BC, Berman-Booty LD, Guild KJ, Du Y, et al. Epithelial-cell-intrinsic IKK-beta expression regulates intestinal immune homeostasis. Nature. 2007 Mar 29;446(7135):552-6.
151. Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol. 2009 Jun;9(6):393-407.
152. Miterski B, Böhringer S, Klein W, Sindern E, Haupts M, Schimrigk S, et al. Inhibitors in the NFkappaB cascade comprise prime candidate genes predisposing to multiple sclerosis, especially in selected combinations. Genes Immun. 2002 Jun;3(4):211-9.
153. Greve B, Weissert R, Hamdi N, Bettelli E, Sobel RA, Coyle A, et al. I kappa B kinase 2/beta deficiency controls expansion of autoreactive T cells and suppresses experimental autoimmune encephalomyelitis. J Immunol. 2007 Jul 1;179(1):179-85.
154. Brüstle A, Brenner D, Knobbe CB, Lang PA, Virtanen C, Hershenfield BM, et al. The NF-κB regulator MALT1 determines the encephalitogenic potential of Th17 cells. J Clin Invest. 2012 Dec;122(12):4698-709.
155. Ruan Q, Kameswaran V, Zhang Y, Zheng S, Sun J, Wang J, et al. The Th17 immune response is controlled by the Rel-RORγ-RORγ T transcriptional axis. J Exp Med. 2011 Oct 24;208(11):2321-33.
156. Yu J, Wang Y, Yan F, Zhang P, Li H, Zhao H, et al. Noncanonical NF-κB activation mediates STAT3-stimulated IDO upregulation in myeloid-derived suppressor cells in breast cancer. J Immunol. 2014 Sep 1;193(5):2574-86.
157. Ellrichmann G, Thöne J, Lee DH, Rupec RA, Gold R, Linker RA. Constitutive activity of NF-kappa B in myeloid cells drives pathogenicity of monocytes and macrophages during autoimmune neuroinflammation. J Neuroinflammation. 2012 Jan 20;9:15.
158. Lee MJ, Bing SJ, Choi J, Jang M, Lee G, Lee H, et al. IKKβ-mediated inflammatory myeloid cell activation exacerbates experimental autoimmune encephalomyelitis by potentiating Th1/Th17 cell activation and compromising blood brain barrier. Mol Neurodegener. 2016 Jul 22;11(1):54.
159. van Loo G, De Lorenzi R, Schmidt H, Huth M, Mildner A, Schmidt-Supprian M, et al. Inhibition of transcription factor NF-kappaB in the central nervous system ameliorates autoimmune encephalomyelitis in mice. Nat Immunol. 2006 Sep;7(9):954-61.
160. Brambilla R, Dvoriantchikova G, Barakat D, Ivanov D, Bethea JR, Shestopalov VI. Transgenic inhibition of astroglial NF-κB protects from optic nerve damage and retinal ganglion cell loss in experimental optic neuritis. J Neuroinflammation. 2012 Sep 10;9:213.
161. Brambilla R, Persaud T, Hu X, Karmally S, Shestopalov VI, Dvoriantchikova G, et al. Transgenic inhibition of astroglial NF-kappa B improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J Immunol. 2009 Mar 1;182(5):2628-40.
162. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011 May 19;473(7347):317-25.
163. Yu XH, Zheng XL, Tang CK. Nuclear Factor-κB Activation as a Pathological Mechanism of Lipid Metabolism and Atherosclerosis. Adv Clin Chem. 2015;70:1-30.
164. Monaco C, Andreakos E, Kiriakidis S, Mauri C, Bicknell C, Foxwell B, et al. Canonical pathway of nuclear factor kappa B activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis. Proc Natl Acad Sci U S A. 2004 Apr 13;101(15):5634-9.
165. Kempe S, Kestler H, Lasar A, Wirth T. NF-kappaB controls the global pro-inflammatory response in endothelial cells: evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005 Sep 21;33(16):5308-19.
166. Gareus R, Kotsaki E, Xanthoulea S, van der Made I, Gijbels MJ, Kardakaris R, et al. Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis. Cell Metab. 2008 Nov;8(5):372-83.
167. Park SH, Sui Y, Gizard F, Xu J, Rios-Pilier J, Helsley RN, et al. Myeloid-specific IκB kinase β deficiency decreases atherosclerosis in low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2012 Dec;32(12):2869-76.
168. Kanters E, Pasparakis M, Gijbels MJ, Vergouwe MN, Partouns-Hendriks I, Fijneman RJ, et al. Inhibition of NF-kappaB activation in macrophages increases atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2003 Oct;112(8):1176-85.
169. Lin Y, Bai L, Chen W, Xu S. The NF-kappaB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin Ther Targets. 2010 Jan;14(1):45-55.
170. Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. 1998 Nov 5;396(6706):77-80.
171. Dudek SE, Luig C, Pauli EK, Schubert U, Ludwig S. The clinically approved proteasome inhibitor PS-341 efficiently blocks influenza A virus and vesicular stomatitis virus propagation by establishing an antiviral state. J Virol. 2010 Sep;84(18):9439-51.
172. Gilmore TD, Herscovitch M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene. 2006 Oct 30;25(51):6887-99.