Abstract
Type II collagen (COL2A1) is the principal structural protein of cartilage and is crucial for maintaining extracellular matrix integrity during skeletal development. Pathogenic variants in COL2A1 disrupt collagen folding, secretion, and fibrillar assembly, leading to defective cartilage architecture and impaired chondrocyte maturation. These molecular alterations underline a broad group of autosomal dominant skeletal disorders collectively known as type II collagenopathies. This review summarizes the structure and regulatory mechanisms of COL2A1, highlighting how specific mutations affect chondrocyte organization and extracellular matrix homeostasis. The discussion also integrates findings from induced pluripotent stem cell (iPSC) based models, which provide patient-specific systems to study the pathogenic consequences of COL2A1 mutations under controlled conditions. Moreover, advances in CRISPR/Cas9-mediated gene editing have enabled the creation of isogenic iPSC lines that replicate or correct pathogenic alleles, offering new opportunities to investigate disease mechanisms and explore targeted therapeutic repair. Collectively, these insights connect gene-level alterations to cellular dysfunction, advancing the understanding of cartilage development and skeletal dysplasia pathogenesis.
Keywords
COL2A1, Type II collagen, Chondrocyte differentiation, Skeletal dysplasia, iPSC, CRISPR/Cas9, Cartilage matrix, Gene editing
Introduction
Skeletal development depends on the precise coordination of cellular differentiation and extracellular matrix (ECM) formation, processes that are fundamental to understanding stem cell-based models of cartilage biology. Endochondral ossification is the developmental process through which cartilage is replaced by bone, forming the long bones of the skeleton. This begins with the condensation of mesenchymal stem cells (MSCs), followed by chondrogenic differentiation and the development of a cartilage template [1]. Within this cartilage template, chondrocytes play a pivotal role as they are responsible for synthesizing, organizing, and maintaining the ECM of the cartilage [1]. The ECM is primarily composed of structural proteins such as collagens and proteoglycans. Type II collagen is the predominant form, accounting for over 90% of the total collagen content in cartilage. It forms a fibrillar network that is intricately associated with proteoglycan aggregates, providing essential tensile strength and mechanical integrity to the tissue [1,2]. Type II collagen is expressed as a homotrimeric protein composed of three α1(II) chains and is synthesized exclusively by chondrocytes [1]. Advances in iPSC technologies now enable researchers to model these pathogenic mechanisms in vitro, providing new opportunities to study collagen type II alpha 1 chain (COL2A1) associated cartilage disorders and test targeted repair strategies [3].
The following section describes the COL2A1 gene’s structure and biological significance.
The COL2A1 gene and type II collagen
Structure and function of COL2A1
Genomic location and organization: The COL2A1 gene encodes the alpha-1 chain of type II collagen, a fibrillar collagen localized to cartilage and the vitreous humor; gene annotations further indicate roles in collagen fibril organization and in conferring tensile strength to the extracellular matrix [4]. To understand how mutations in this gene affect skeletal development, it is important to begin with its genomic context. Genomic mapping places COL2A1 on chromosome 12q13, with earlier studies mapping it specifically to 12q13.1-q13.2 and reporting a gene span of approximately 31.5 kb [5]. The NCBI RefSeq gene summary lists the locus as 12q13.11 and reports an exon count of 57, whereas other reports describe COL2A1 as a 54-exon gene [5,6]. In line with these annotations, the canonical COL2A1 transcript NM_001844.4 is reported to contain 54 exons and to span roughly 31.5 kb [5,6]. The COL2A1 gene has been annotated differently across major genomic databases. According to the NCBI Gene record, COL2A1 is located at 12q13.11 and consists of 57 exons [4], while multiple peer-reviewed studies describe it as a 54-exon gene spanning approximately 31.5 kb on chromosome 12q13.11 [5–8] .This inconsistency highlights the importance of specifying transcript versions when reporting variants to ensure accurate genomic mapping. However, the available literature does not clearly address whether these discrepancies arise from differences in annotation methods, transcript models, or alternative splicing. Furthermore, transcript diversity arises through alternative splicing. For example, a splice-junction variant was shown to cause aberrant mRNA splicing that produced an alternative, shorter COL2A1 transcript via complete skipping of exon 10 (c.655_708del), resulting in an 18-amino-acid deletion (p.Gln221_Pro238del). The authors note that splice-affecting variants can lead either to nonsense-mediated decay or to the production of an in-frame protein with a small deletion [9]. Regulation is also mediated by conserved intronic sequences. Among these, a well-studied SRY-box transcription factor 9 (SOX9) dependent enhancer in intron 1 and another SOX9-bound region within intron 6 are essential for transcriptional activation. Functional assays indicate that a short sequence within intron 6 is required for COL2A1 expression and for SOX9 binding in chondrocyte cells [10].
Building on this, genome-wide enhancer mapping in fetal chondrocytes identified thousands of chondrocyte active enhancers and demonstrated that experimentally deleting enhancers at the COL2A1 locus alters gene expression, thereby validating their role in chondrocyte regulatory specificity [11].
Transcriptional regulation: Building on its genomic organization, COL2A1 expression is tightly controlled at the transcriptional level. In particular, SOX9 acts as the master regulator of COL2A1, functioning together with SOX5 and SOX6 to enhance transcription. These transcription factors act through both the intron-1 enhancer and the intron-6 enhancer [10]. Genome-wide mapping of chondrocyte enhancers confirmed this model, identifying thousands of putative chondrogenic enhancers enriched for SOX9 and other lineage-specific motifs (such as myocyte enhancer factor-2 (MEF2) and forkhead box (FOX). Targeted deletions at the COL2A1 locus further demonstrated that loss of specific enhancers reduces COL2A1 transcript levels, highlighting their functional importance [11].
In addition to transcription factors, epigenetic mechanisms enable proper regulation. For example, ten-eleven translocation methylcytosine dioxygenase 1 (TET1)-mediated hydroxymethylation (5-hydroxymethylcytosine, 5hmC) accumulates at SOX9 peaks during chondrogenesis, promoting SOX9 binding and supporting activation of targets such as COL2A1 and ACAN. Loss of TET1 reduces SOX9 target activation despite unchanged SOX9 levels, underscoring the importance of epigenetic remodeling in this process [12].
Protein domains of type II collagen: At the protein level, COL2A1 encodes the procollagen α1(II) chain of roughly 1,487 amino acids. This chain is synthesized with N- and C-terminal propeptides and folds into a triple-helical structure characterized by the Gly-X-Y repeat motif, where glycine occupies every third position [4,5]. The presence of glycine at these positions is essential for correct helix folding and stability, and substitutions of glycine within the triple helix represent a predominant COL2A1 mutation type that can severely impair collagen assembly [5]. The C-terminal non-collagenous (C-propeptide) domain is critical for chain association and triple-helix nucleation. Pathogenic changes in this domain can disrupt collagen assembly and secretion, thereby reducing extracellular fibril deposition and contributing to chondrodysplasia [5]. In fact, problems in processing chondrocalcin, the C-propeptide, have been directly linked to skeletal abnormalities. After biosynthesis, the procollagen undergoes extensive post-translational processing, including cleavage of the N- and C-propeptides and cross-linking, to generate mature type II collagen fibrils. These fibrils are integral to cartilage extracellular matrix structure, where they confer tensile strength and support tissue function [4].
Role of type II collagen in cartilage and skeletal development
Cartilage structure and extracellular matrix integrity: COL2A1 consists of three identical α1(II) chains that assemble into a triple-helical structure, and this triple-helical molecule confers tensile strength and structural stability to cartilaginous tissues and contributes to cartilage development and the maintenance of joint integrity and function [13]. Within the cartilage extracellular matrix, macromolecular organization is essential for tissue mechanostability across development and adult life [14]. COL2A1 constitutes up to 60% of the extracellular matrix and provides the fibrillar assembly, whereas the extrafibrillar network is composed predominantly of aggrecan bound to hyaluronan. Interactions between these supramolecular assemblies are stabilized by mutual binding to adaptor protein complexes within a perifibrillar compartment [14,15].
Interactions with other collagens and extracellular matrix (ECM) proteins that stabilize fibrils: In addition to its intrinsic structural role, type II collagen engages in multiple interactions with other collagens and extracellular matrix proteins that ensure fibril stability. Type II collagen fibrils are laterally associated with minor fibrillar and FACIT collagens, notably types IX and XI, which together stabilize fibril architecture and regulate fibril diameter, while adaptor proteins, including cartilage oligomeric matrix protein (COMP) and matrilins, form perifibrillar complexes that further stabilize the matrix [14,16]. COL2A1 occurs in mixed fibrils with collagens IX and XI in cartilage [16]. Collagen IX is proposed to stabilize the fibrillar and proteoglycan networks through lateral association with collagen II and collagen XI [14]. Collagen type XI both nucleates fibril formation and regulates fibril diameter, supporting a role for these associated collagens in maintaining fibril architecture and size [16]. Furthermore, an interacting complex composed of collagen IX, matrilin-3, and COMP has been described as essential for cartilage matrix stability, consistent with matrilins and COMP functioning as perifibrillar adaptor proteins that stabilize the extracellular matrix [14]. Disruption of collagen IX-dependent interactions perturbs extracellular matrix organization in developing cartilage [14]. These changes affect chondrocyte survival and disturb normal tissue architecture in the growth plate, resulting in growth retardation and short stature [14]. At later stages, cartilage abnormalities progress into an osteoarthritis-like phenotype, characterized by the depletion of proteoglycans and a reduction in intact COL2A1 [14].
COL2A1 expression in chondrocytes, developmental regulation, and pathological relevance: Beyond structural contributions, the role of COL2A1 is tightly regulated at the gene expression level during chondrogenesis. COL2A1 is an established marker of the chondrogenic phenotype during chondrogenesis, and during early differentiation, mRNA levels of COL2A1, aggrecan (ACAN), and SOX9 increase, while as differentiation progresses, cells change their expression to include upregulation of the hypertrophic chondrocyte markers COL10A1, RUNX2, and Matrix metalloproteinase 13 (MMP13), reflecting hypertrophic maturation in the course of endochondral ossification [16]. Epigenetic and microenvironmental cues influence COL2A1 transcription and collagen II abundance, and for example, three-dimensional culture conditions recruit the protein deacetylase sirtuin-1 (SirT1) and the histone methyltransferase Set7/9 to the COL2A1 promoter, as shown by chromatin immunoprecipitation analyses, and these events are associated with augmented COL2A1 expression, with the data indicating that Set7/9 can locally repress SirT1 enzymatic activity at the promoter leading to increased COL2A1 expression in a cell morphology dependent manner [17]. COL2A1 mutations that destabilize the collagen triple helix or impair secretion have been implicated in severe type II collagenopathies such as perinatal-lethal achondrogenesis type II (ACG2) [13]. MMP13, which is upregulated during late stages of chondrogenic differentiation, degrades major extracellular matrix components, including aggrecan and type II collagen, and thus mediates extracellular matrix degradation during hypertrophic progression [15]. Perturbation of extracellular matrix composition or homeostasis can disrupt growth-plate organization and chondrocyte survival and, in vivo, has been associated with osteoarthritis-like degeneration characterized by proteoglycan depletion and loss of intact type II collagen, linking dysregulated matrix remodeling to skeletal growth abnormalities and degenerative cartilage phenotypes [14].
Stages of chondrocyte maturation
Chondrocyte maturation proceeds through successive stages from progenitor mesenchymal cells to hypertrophic chondrocytes, preceding terminal cell death. Hypertrophic chondrocytes regulate matrix mineralization and blood vessel invasion, culminating in bone formation by endochondral ossification [18]. These maturation steps proceed in a temporally organized sequence, each stage building upon molecular and structural cues established in the previous one.
Mesenchymal condensation and early chondrogenic commitment: Mesenchymal condensation is a developmental process in which dispersed cells are condensed to high local cell density to differentiate into specific tissue types, and it represents an early stage of selective gene activation in tissue morphogenesis. In vitro platforms that mimic ECM-driven condensation, for example, collagen microencapsulation of human mesenchymal stromal cells (hMSC), reproduce early condensation events, such as ECM-driven, cell-density-dependent volume contraction and increased local cell density. Moreover, collagen microencapsulation of hMSC produces a transient upregulation of condensation markers and an enhanced nuclear localization of SOX9, consistent with potentiation of subsequent chondrogenic differentiation. Proper spatiotemporal expression of cell-cell adhesion molecules, specifically N-cadherin and neural cell adhesion molecule (N-CAM), is required for the condensation process and thereby supports chondrogenesis [19]. Following condensation, the cells enter a proliferative phase that marks the next step of cartilage template formation.
Proliferating chondrocytes: Following condensation, cells transiently exit the cell cycle and enter chondrogenesis by upregulating SOX9. Chondroblasts subsequently resume proliferation and contribute to the formation of the cartilage anlage [18]. SOX9 expression increases are observed at the prechondrocyte stage while COL2A1 mRNA levels remain low, with COL2A1 upregulation and the first signs of collagen II matrix deposition becoming evident as chondroblasts proliferate and mature [18]. In developmentally inspired in vitro systems, sustained SOX9 activity combined with chondro-inductive stimulation (for example, transforming growth factor-β3, TGF-β3) maintains SOX9 expression and results in robust expression and extracellular deposition of ACAN and COL2A1, indicating successful chondrogenesis and formation of cartilage-like ECM [18,19]. As chondrocytes continue to proliferate, they gradually transition toward a pre-hypertrophic state characterized by key molecular shifts.
Pre-hypertrophic transition: SOX9 is expressed in resting, proliferative, and pre-hypertrophic growth-plate chondrocytes but is substantially downregulated approximately fivefold in the hypertrophic zone [19]. Indian hedgehog (IHH) expression increases as chondrocytes enter the hypertrophic program, concurrent with upregulation of hypertrophy-associated markers [18]. RUNX2 is a critical regulator of chondrocyte hypertrophy and directly regulates IHH expression further. RUNX2 can functionally cooperate with cofactors such as doublesex and mab-3 related transcription factor 2 (DMRT2) to enhance IHH and other hypertrophic gene expression in chondrocytes [20]. COL10A1 expression begins to rise with the onset of hypertrophic differentiation, marking the early stages of matrix remodeling and cellular enlargement that characterize the hypertrophic program [18,20]. These regulatory events collectively prepare the chondrocytes for full hypertrophic differentiation.
Hypertrophic chondrocytes: Hypertrophic chondrocytes are characterized by increased cell volume and by upregulation of hypertrophic markers such as COL10A1 and alkaline phosphatase (ALPL). These late-stage chondrocytes produce MMP13, which facilitates vascular invasion as the ECM becomes mineralized before replacement by bone via endochondral ossification [18]. DMRT2 is selectively expressed in pre-hypertrophic chondrocytes and collaborates with RUNX2 to synergistically induce RUNX2 target genes, including COL10A1 and ALPL, thereby promoting hypertrophic gene expression and the matrix changes that precede cartilage resorption and ossification [18,20]. Once hypertrophic differentiation is achieved, chondrocytes progress toward terminal maturation and eventually fate determination.
Terminal maturation and apoptosis: The final fate of terminal hypertrophic chondrocytes is not uniformly apoptotic, because lineage-tracing and genetic experiments demonstrate that a substantial subset of hypertrophic chondrocytes can transdifferentiate into osteoblast-lineage cells and contribute to trabecular and cortical bone formation [21]. SOX9 expression is physiologically downregulated in the hypertrophic zone of the growth plate, and osteoblast-associated genes such as MMP13, COL1A1, and integrin-binding sialoprotein (IBSP) are normally upregulated beginning in the lower hypertrophic zone as cells prepare to enter bone-forming programs [19]. Experimental maintenance of SOX9 expression in chondrocyte-lineage cells suppressed the normal upregulation of these osteoblastic genes and substantially reduced the number of chondrocyte-derived cells that appear in the trabecular bone, and persistent SOX9 expression was associated with increased markers of apoptosis in terminal hypertrophic chondrocytes [19]. Conversely, zinc finger and BTB domain containing 20 (ZBTB20) represses SOX9 in late hypertrophic chondrocytes, and chondrocyte-specific deletion of ZBTB20 causes ectopic SOX9 expression in the late hypertrophic zone, decreased vascular endothelial growth factor A (VEGFA) expression, delayed cartilage vascularization, expansion of the hypertrophic zone, and delayed endochondral ossification [13]. This sequential progression through maturation stages is schematically illustrated (see Figure 1) [22].
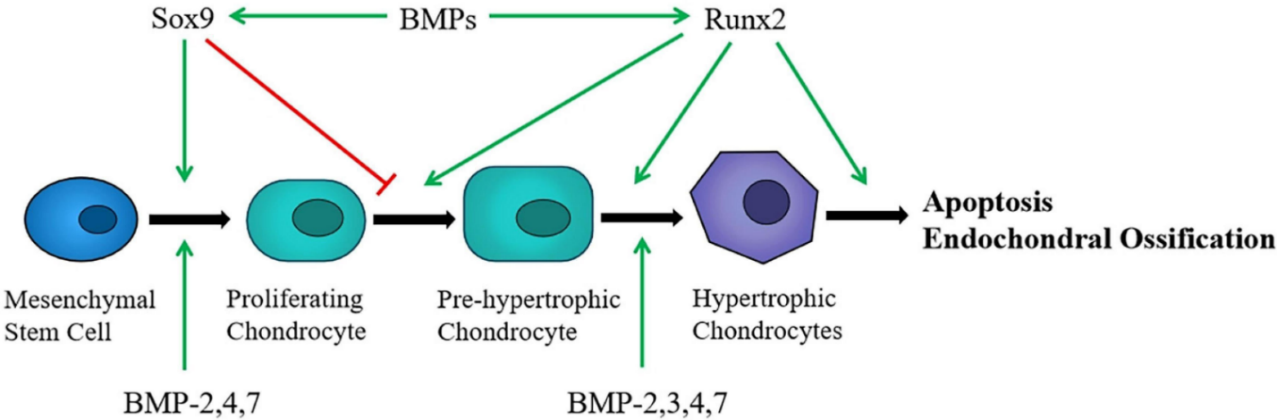
Figure 1. Schematic summary of chondrocyte maturation stages from mesenchymal stem cells to terminal differentiation, highlighting key regulators SOX9, RUNX2, and BMP signaling. Adapted from Chen et al. (2021) [22].
Regulatory pathways involved in chondrogenesis
Multiple signal transduction pathways regulate chondrocyte differentiation and together form a complex transcriptional network whose balanced activity is essential for normal chondrocyte differentiation and cartilage development [23,24].
SOX trio (SOX5, SOX6, SOX9): The SOX transcription factor trio SOX5, SOX6, and SOX9 ranks highly within these networks and functions as core regulators of the chondrogenic program [25]. SOX9 in particular is the earliest nuclear factor required for chondrogenesis and is indispensable to establishing the chondrogenic lineage [23]. SOX5 and SOX6 are closely related DNA-binding proteins that critically enhance SOX9 function and thereby cooperate with SOX9 to activate chondrocyte-specific transcriptional programs [25]. Expression of the three SOX genes peaks in proliferating and pre-hypertrophic growth plate chondrocytes and is abruptly turned off when chondrocytes undergo hypertrophy [25]. Genome-wide analyses indicate that the SOX proteins primarily target enhancers and frequently occupy super-enhancers associated with cartilage-specific genes, and the SOX trio directly controls genes encoding major components of the cartilage ECM, including COL2A1 [25]. Collectively, these transcription factors establish the molecular framework for chondrogenesis, which is subsequently modulated by signaling pathways such as Indian hedgehog (Ihh)–parathyroid hormone-related protein (PTHrP).
Indian hedgehog (ihh) and parathyroid hormone-related protein (pthrp) loop: Ihh is expressed and secreted by pre-hypertrophic and early hypertrophic chondrocytes and functions as a key regulator of endochondral ossification by directly promoting proliferation of proliferating chondrocytes and by upregulating PTHrP, thereby establishing a negative-feedback loop that restrains premature chondrocyte hypertrophy [23,24]. PTHrP signals via the PTH/PTHrP receptor (PTHR1) to activate Gαs-mediated adenylate cyclase → cyclic adenosine monophosphate (cAMP) → protein kinase A signaling, which maintains chondrocyte proliferation and promotes activation of SOX9, thereby delaying hypertrophic differentiation. In contrast, Gαq-mediated phospholipase C → inositol trisphosphate/diacylglycerol signaling through the same receptor can favor progression toward hypertrophy, so the overall effect depends on pathway coupling and cellular context [23,24]. The feedback mechanisms of Ihh–PTHrP signaling closely integrate with morphogenetic factors like bone morphogenetic proteins (BMPs) to coordinate proliferation and hypertrophic timing.
Bone morphogenetic proteins (BMPs): BMPs are positive regulators of ectopic chondrogenesis and endochondral ossification, acting through BMP type I and II receptors to phosphorylate receptor-SMADs (SMAD1, SMAD5, and SMAD8/9), a signaling axis that contributes to chondrogenic gene regulation including effects on SOX9 and cartilage matrix formation [24]. Comparative in vitro data show that articular chondrocyte cultures enter differentiation with higher early BMP4 expression, constitutive phosphorylated SMAD1/5/9 signaling and elevated SOX9 protein levels that correspond to faster chondrogenic re-differentiation, and experimental BMP-4/7 co-treatment produces additive increases in SOX9 consistent with a pro-chondrogenic role of canonical BMP signaling during MSC chondrogenesis [24,26]. Nevertheless, pharmacologic silencing of canonical BMP–SMAD1/5/9 signaling (for example with the BMP-receptor inhibitor dorsomorphin) reduced SOX9, altered RUNX2 and alkaline phosphatase responses, and overall decelerated MSC differentiation rather than selectively blocking hypertrophy, implying that SOX9 induction can depend on BMP/SMAD inputs and that BMP inhibition may slow differentiation instead of uncoupling matrix production from hypertrophic progression [26].
WNT/β-catenin signaling: Canonical WNT signaling via β-catenin promotes osteoblast differentiation and concurrently suppresses chondrogenesis, as activation of WNT favors osteoblastogenesis over cartilage formation, and activation of the canonical WNT/β-catenin pathway has been shown to block early chondrogenic differentiation from MSCs while promoting chondrocyte hypertrophy [24]. By contrast, noncanonical WNT branches, including WNT5A and WNT5B acting through planar cell polarity signaling, play distinct roles in regulating chondrocyte proliferation and growth-plate columnar morphogenesis rather than driving the canonical hypertrophic program. Therefore, appropriate regulation of WNT signaling levels is required for normal transitions in chondrocyte maturation, because different WNT ligands and pathways have divergent effects on proliferation, differentiation, and hypertrophy [23]. This WNT-dependent regulation is further refined by fibroblast growth factor (FGF) signaling, which modulates cell behavior in a stage-dependent manner.
Fibroblast growth factors (FGFs): FGF ligands acting through fibroblast growth factor receptors (FGFRs) regulate chondrocyte behavior in a stage-dependent manner, promoting chondrocyte proliferation and differentiation during early development but inhibiting chondrocyte proliferation and differentiation at later developmental stages [23]. FGFR3 is expressed predominantly in proliferating chondrocytes in the growth plate, where it regulates cell growth and differentiation [23,24]. Activation of FGFR3 limits chondrocyte proliferation, and while FGFR3 signaling has been described to inhibit proliferation and hypertrophy in later stages, activating FGFR3 mutations have been reported to inhibit the initiation of chondrocyte hypertrophy yet accelerate late hypertrophic differentiation [23,24]. Clinically relevant gain-of-function mutations in FGFR3 cause achondroplasia and related chondrodysplasia syndromes by producing constitutive overactivation of FGF signaling, which reduces chondrocyte proliferation and impairs hypertrophic differentiation in the growth plate, producing the characteristic growth-plate and short-limb phenotypes [23]. These growth factor pathways converge on transcriptional regulators, including RUNX2 and myocyte enhancer factor-2C (MEF2C), to drive hypertrophic gene expression and matrix remodeling.
RUNX2 and MEF2C: The Runt-family transcription factors RUNX2 and RUNX3 are expressed in pre-hypertrophic and hypertrophic chondrocytes and are regarded as important drivers of endochondral differentiation [24]. MEF2C is likewise critical for early-stage chondrocyte hypertrophy, and chondrocyte-specific loss of MEF2C in mice results in suppression of hypertrophy and impaired endochondral ossification [27]. MEF2C has been reported to act upstream of RUNX2 and to regulate COL10A1 expression, linking MEF2C activity to downstream hypertrophic marker induction [24,27]. Ectopic expression of RUNX2 in immature chondrocytes induces hypertrophic marker genes including COL10A1 and MMP13, consistent with a role for Runt factors in driving cartilage-to-bone transitional gene expression [24]. In human mesenchymal progenitor cell chondrogenesis in vitro, Dreher and colleagues observed that BMP signaling stimulated RUNX3 expression, and the same study found that FGF receptor signaling modulated RUNX2 expression via ERK activity, as FGFR inhibition reduced phosphorylated ERK1/2 and RUNX2 levels. Meanwhile, MEF2C abundance was selectively affected by manipulations of WNT signaling and pulsed PTHrP/hedgehog treatments—interventions that were the most potent anti-hypertrophic measures in their model—indicating that MEF2C regulation differs from that of RUNX2 and RUNX3. Together, these pathway-specific dependencies indicate non-redundant regulation of RUNX2, RUNX3, and MEF2C during human MSC chondrogenesis and support the interpretation that RUNX2, on the one hand, and RUNX3/MEF2C, on the other, have distinct roles in steering progenitor cells toward endochondral versus alternative lineage outcomes [27]. As shown in Figure 2, the TGFβ, BMP, and FGF pathways converge on proliferating, pre-hypertrophic, and hypertrophic chondrocytes to coordinate stage-dependent regulation of chondrogenesis.
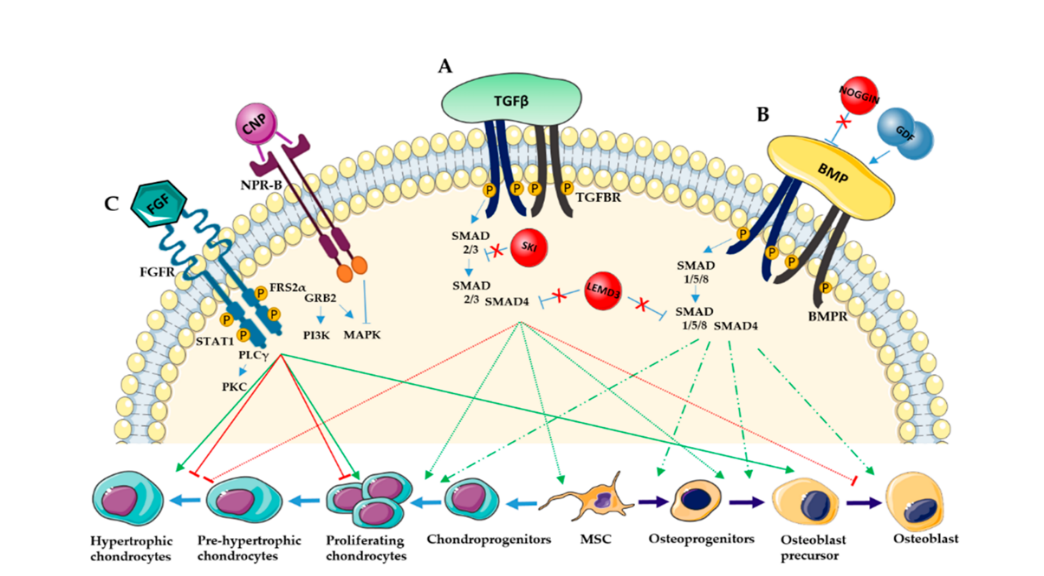
Figure 2. Key signaling pathways (TGFβ, BMP, and FGF) regulating chondrocyte proliferation, differentiation, and hypertrophy through SMAD and MAPK cascades. Adapted from Guasto et al. (2021) [23].
Cellular and molecular alterations in cartilage
Chondrocyte phenotype loss (dedifferentiation): Adult articular chondrocytes undergo senescence and dedifferentiation during prolonged in vitro expansion, leading to loss of their specialized phenotype and restricting successful cartilage regeneration. Late passage chondrocytes display reduced expression of COL2A1 and decreased proteoglycan content, together with increased expression of hypertrophic markers such as COL10A1, consistent with a shift toward a dedifferentiated/hypertrophic state. These phenotypic and molecular alterations typically appear after serial passaging (notably after the fourth passage) and are associated with a reduced capacity to regenerate hyaline cartilage and, when transplanted, can produce undesirable fibrous repair tissue [28].
Cellular senescence and apoptosis (SASP, mitochondria, oxidative stress): Articular cartilage in osteoarthritis exhibits markedly increased numbers of senescent chondrocytes, with the proportion of cells positive for p16INK4a, p21, and p53 rising notably in destabilization of the medial meniscus (DMM) models and diseased cartilage [29]. Senescent chondrocytes express a senescence-associated secretory phenotype (SASP), defined as the production of proinflammatory cytokines, chemokines, growth factors, and proteolytic enzymes. These SASP factors, including matrix metalloproteinases, have been linked experimentally to ECM degradation, and interventions that reduce SASP and MMP expression mitigate IL-1β-driven loss of COL2A1 and aggrecan in cartilage models. Consistent with these observations, IL-1β is widely used as an in vitro inflammatory stimulus because it induces chondrocyte apoptosis and senescence and provokes SASP expression [30]. Mechanistically, perturbations of mitochondrial homeostasis are implicated in chondrocyte senescence induced by ECM stiffening in mechanically stressed cartilage. Experimental increases in ECM stiffness elevate mitophagy in chondrocytes and are accompanied by upregulation of mitophagy markers including PINK1, Parkin, and LC3 in cells cultured on higher stiffness substrates. Pharmacologic or genetic inhibition of Parkin-dependent mitophagy attenuates the senescent phenotype and reduces senescence marker expression in chondrocytes and in mouse joints, supporting a role for altered mitochondrial turnover in promoting chondrocyte senescence under stiffness stress [29].
Extracellular matrix (ECM) degradation (catabolic enzymes, proteoglycan and collagen loss): Inflammatory and degenerative stimuli increase expression of matrix-degrading enzymes and reduce anabolic ECM components in chondrocytes [30, 31]. IL-1β stimulation of chondrocytes leads to elevated expression of MMP13 and concomitant suppression of COL2A1 expression [31]. Therapeutic interventions that attenuate IL-1β-driven SASP signaling restore levels of COL2A1 and aggrecan in vitro, as exemplified by ononin and procyanidin B2 [30,31]. Macroscopically, osteoarthritic cartilage exhibits increased matrix stiffness together with evident surface and structural defects that are positively correlated with progressive cartilage matrix deterioration [29].
Inflammatory mediators and signaling (IL-1β, TNF-α, IL-6; NF-κB/MAPK): Proinflammatory cytokines and chemokines produced in osteoarthritic cartilage, sub-chondral bone, and synovium drive the release of MMPs, notably MMP13, thereby promoting ECM catabolism and cartilage degradation [31]. IL-1β stimulation markedly increases production of the inflammatory mediators TNF-α and IL-6 by chondrocytes [31]. The MAPK family—including ERK, JNK, and p38—regulates cellular processes such as survival, apoptosis, proliferation, and inflammatory responses and is activated in this context [31]. Activation of MAPK, together with NF-κB signaling, underpins the sustained inflammatory and matrix-degrading program in osteoarthritic cartilage. NF-κB activation is necessary for chondrocyte expression of MMPs and exacerbation of cartilage destruction [31].
Epigenetic and metabolic reprogramming: Epigenetic mechanisms modulate articular chondrocyte regeneration and age-related disease processes. Genome-wide DNA methylation patterns and transcriptional regulators such as STAT3 shape chondrocyte epigenetic state and biological age, as evidenced by an epigenetic clock that links CpG methylation to chondrocyte age and by the observation that pharmacologic STAT3 activation lowers epigenetic age in adult chondrocytes. Chondrocyte function and regenerative capacity decline with advancing chronological and epigenetic age [32]. Moreover, epigenetic regulators such as HDAC3 have been identified as mechanosensitive mediators. Matrix stiffening downregulates HDAC3, which in turn promotes PINK1/Parkin-dependent mitophagy and stimulates chondrocyte senescence and osteoarthritis progression, indicating that such epigenetic factors transduce pathological mechanical stimuli into altered chondrocyte phenotypes [29].
Mechanical and biomechanical stress (mechanotransduction, integrin/ERK signaling): Abnormal biomechanical environments alter chondrocyte behavior through mechanotransductive pathways. ECM stiffening is a characteristic feature of cartilage aging and osteoarthritis and is observed as increased matrix stiffness in diseased cartilage [29]. ECM stiffening promotes phenotypic deterioration of chondrocytes via mechanosensitive epigenetic regulation, with HDAC3 identified as a key mechanosensitive regulator whose downregulation by matrix stiffening drives downstream signaling that enhances Parkin acetylation, activates PINK1/Parkin-dependent mitophagy, and stimulates chondrocyte senescence and cartilage degeneration [29]. Dynamic loading and shear have been shown to modify chondrocyte gene expression and to activate MAPK signaling, including the MEK/ERK axis. Degenerative stimuli are associated with downregulation of the chondrogenic transcription factor SOX9 [28,29].
Pathogenic variants and molecular consequences in COL2A1
The COL2A1 gene encodes the alpha 1 chain of type II collagen (COL2A1). Molecular defects in COL2A1 give rise to a wide spectrum of autosomal dominant type II collagenopathies affecting connective tissues and the skeleton. Many pathogenic COL2A1 variants are missense substitutions clustered in the triple helical Gly X Y repeat, most notably glycine substitutions and arginine to cysteine changes, which frequently exert dominant negative effects [33]. Mutant type II collagen proteins often show reduced thermostability, delayed secretion, and intracellular retention, reflecting impaired folding and post translational maturation. These intracellular defects, together with an abnormal extracellular collagenous matrix, perturb growth plate organization and underlie the skeletal phenotypes [33,34].
Mutation-sensitive regions of COL2A1: Type II collagen (COL2A1) possesses a large, uninterrupted triple helical domain composed of Gly X Y repeats that constitutes the predominant locus of pathogenic substitutions. Within this triple helical region, glycine substitutions are particularly frequent, and arginine to cysteine substitutions account for a smaller but notable proportion of variants [33]. Mutations involving the C propeptide region have been reported, and dominant negative alterations within this domain have been linked to severe phenotypes, including platyspondylic lethal skeletal dysplasia (Torrance type) [33,34].
Functional consequences of mutations: Substitutions in the triple helical Gly X Y repeats of type II collagen (COL2A1), particularly glycine substitutions, are commonly reported and are associated with disease related alterations in the mutant molecules [33]. Mutations in the C propeptide region of the COL2A1 α1 chain have also been described and, in experimental models, correspond to intracellular accumulation of the mutant protein within expanded rough endoplasmic reticulum and induction of endoplasmic reticulum stress markers [33,34]. Consistently, experimental models demonstrated that mutant type II collagen molecules show altered electrophoretic mobility, relatively low thermostability, and slow rates of secretion into the extracellular space [33]. By contrast, for collagen type X (COL10A1), many reported NC1 domain mutations impair trimerization: mutant chains "did not form stable homotrimers and were unable to form stable heterotrimers with wild type chains," a defect that is accompanied by intracellular retention and activation of the unfolded protein response [34]. Notably, some pathogenic variants (for example Arg→Cys substitutions) can evade or fail to engage canonical ER proteostasis surveillance, allowing malformed procollagen to persist and be secreted or partially retained, with consequent deposition of a structurally compromised matrix [35].
Collagen assembly and chondrocyte impact: COL2A1 mutants deposit a defective, and notably sparser, collagen II matrix in cartilage. Slowed folding of mutant procollagen can produce excess post-translational modification that "could still contribute to defects in fibril formation and crosslinking," yielding impaired fibrillogenesis and altered matrix crosslinking [35]. Mutant collagen fibrils also "self-assemble in abnormal [forms] which are not able to properly interact with other elements of the extracellular fibrillar matrix," thereby disrupting key extracellular matrix interactions [33]. At the tissue level, intracellular retention of mutant collagen in expanded rough endoplasmic reticulum induces endoplasmic reticulum stress and activation of the unfolded protein response. This stress response is associated with an "aberrant columnar organization of chondrocytes" together with reduced proliferation in growth plate chondrocytes, indicating impaired proliferation and differentiation within the growth plate [34]
Classification of COL2A1 mutations
Mutations in genes encoding fibrillar collagens, such as the Type I collagen gene (COL1A1), exhibit a wide mutational spectrum and substantial genetic and clinical heterogeneity [36]. The resulting clinical phenotype frequently depends on mutation class and, in some cases, on the position of the variant within the protein sequence [36,37]. In studies of collagen disorders, genotypes are commonly categorized into two principal mechanisms:
- Haploinsufficiency or quantitative defects (for example, nonsense, frameshift, or whole-gene deletions that reduce collagen amount).
- Structural or qualitative defects (for example, glycine substitutions in the Gly–X–Y triple-helix) [36,37].
These qualitative versus quantitative classifications help to explain observed differences in clinical severity, with structural (qualitative) defects typically associated with more severe phenotypes than quantitative defects [36,37].
Missense mutations: Missense substitutions that replace glycine residues within the Gly-X-Y triplet of the collagen triple helix commonly produce structurally abnormal collagen molecules and are a frequent class of disease-causing variants in fibrillar collagens [38]. Moreover, the substitution of glycine by other amino acids in the Gly-X-Y triplet is described in the literature as a major cause of structurally abnormal collagen synthesis and is noted as one of the principal molecular mechanisms underlying fibrillar collagen disorders [37]. Consistently, glycine substitutions of this kind are classically associated with a dominant-negative effect and are therefore linked to more severe clinical phenotypes than haploinsufficiency mutations [37,39].
Nonsense mutations: In contrast, nonsense variants, together with many frameshift changes and canonical splice-site mutations, commonly introduce premature termination codons and trigger degradation of the corresponding transcript via nonsense-mediated mRNA decay or produce severely truncated, unstable polypeptides. In Type I collagen (COL1A1), these types of variants are classified as quantitative (haploinsufficiency) mutations that reduce the amount of normally structured procollagen chains available for trimer assembly [37,38]. Because haploinsufficiency causes a quantitative deficit of structurally normal procollagen rather than the incorporation of aberrant α-chains into collagen trimers, it is generally associated with milder skeletal phenotypes than the dominant-negative effects produced by structural glycine substitutions in the triple helix [36,38].
Splice-site mutations: On the other hand, splice-site variants show heterogeneous molecular outcomes. In some cases, splice-site lesions cause nonsense-mediated decay (NMD) of the aberrant transcript, producing a quantitative reduction in gene product consistent with haploinsufficiency [37]. Conversely, splice-site alterations can disrupt pre-mRNA processing and generate aberrant transcripts that evade nonsense-mediated decay. Such transcripts may produce altered polypeptides that act in a dominant-negative fashion and are associated with more severe clinical phenotypes [40].
Frameshift mutations: Similarly, frameshift mutations produce haploinsufficiency by causing failure to synthesize the products of one COL1A1 allele [37]. COL1A1 haploinsufficiency produces a quantitative defect, with structurally normal Type I procollagen synthesized at approximately half of the normal amount [38]. These types of mutations can also initiate nonsense-mediated decay (NMD) of the mutant mRNA from the affected allele and are associated with milder bone fragility consistent with haploinsufficiency-type phenotypes, in contrast to structurally disruptive helical glycine substitutions [37,38].
Pathogenic mechanisms
Mutations in COL2A1 produce disease through distinct molecular mechanisms that can be grouped conceptually and that result in overlapping clinical phenotypes, from severe perinatal skeletal dysplasias to milder articular disorders [33,41].
Dominant-negative effects: One major mechanism is dominant-negative disruption. Dominant-negative effects constitute one of the principal molecular mechanisms underlying dominant COL2A1-related disorders, together with haploinsufficiency [41]. Dominant-negative mutations frequently involve substitution of glycine residues within the Gly X Y repeat of the triple-helical domain. These glycine replacements impair homotrimer assembly and destabilize the triple helix, producing abnormal procollagen conformation and reduced thermostability [41]. Mutant α1(II) chains affect assembled procollagen molecules by altering half of the chains in the trimer, yielding unstable trimers with impaired secretion or intracellular accumulation in some cases, and provoking disturbances in extracellular matrix organization and chondrocyte function [34,41]. Well-characterized classes of dominant-negative alterations include glycine substitutions within the triple helix and arginine→cysteine substitutions, both of which have been recurrently observed and are associated with severe and variable skeletal phenotypes [33,41].
Haploinsufficiency: In addition to dominant-negative effects, haploinsufficiency represents another major pathogenic pathway.
Type II collagen (COL2A1) haploinsufficiency arises from truncating variants (nonsense or frameshift) or large deletions that introduce premature termination codons and thereby commonly trigger nonsense-mediated decay (NMD), reducing the synthesis of normal collagen and weakening the articular cartilage matrix [41]. Clinically, haploinsufficiency produces relatively milder manifestations within the type II collagenopathy spectrum. For example, many cases of Stickler syndrome are attributable to mutations that cause reduced COL2A1 dosage rather than dominant-negative structural disruption [41,42].
Endoplasmic reticulum (ER) stress and cellular toxicity: At the cellular level, COL2A1 mutations also trigger endoplasmic reticulum (ER) stress. A subset of type II collagen (COL2A1) missense mutations produces mutant procollagen chains that misfold or fail to assemble properly within the rough ER, resulting in intracellular retention and conspicuous dilation of the ER in affected chondrocytes [33,41]. Accumulation of misfolded collagen activates the unfolded protein response (UPR), an evolutionarily conserved ER stress signaling program. When UPR activation is insufficient to restore proteostasis, pro-apoptotic mediators, including C/EBP homologous protein (CHOP) and caspase pathways, become upregulated, culminating in chondrocyte apoptosis [34,43]. The molecular events underlying UPR activation are outlined in Figure 3, which illustrates how the ER stress sensors—PERK, ATF6, and IRE1—are activated upon accumulation of misfolded proteins. These pathways lead to the induction of transcription factors such as ATF4, cleaved ATF6, and spliced XBP1 (sXBP1), which in turn regulate genes involved in oxidative stress response, ER-associated degradation (ERAD), and apoptosis [42]. In vivo models show that ER stress and UPR in proliferating and/or hypertrophic chondrocytes alter chondrocyte differentiation and perturb growth-plate organization, producing growth plate phenotypes—for example, expansion of the hypertrophic zone and dwarfism—that recapitulate aspects of human metaphyseal and spondyloepiphyseal chondrodysplasias [34,43]. Collectively, these observations indicate that certain COL2A1 mutants act via an intracellular toxic gain-of-function (intracellular proteotoxicity) driven by retention of mutant collagen and ER stress, in addition to any extracellular matrix deficiency. This intracellular toxicity is a major determinant of phenotype severity across multiple experimental models [34,43].
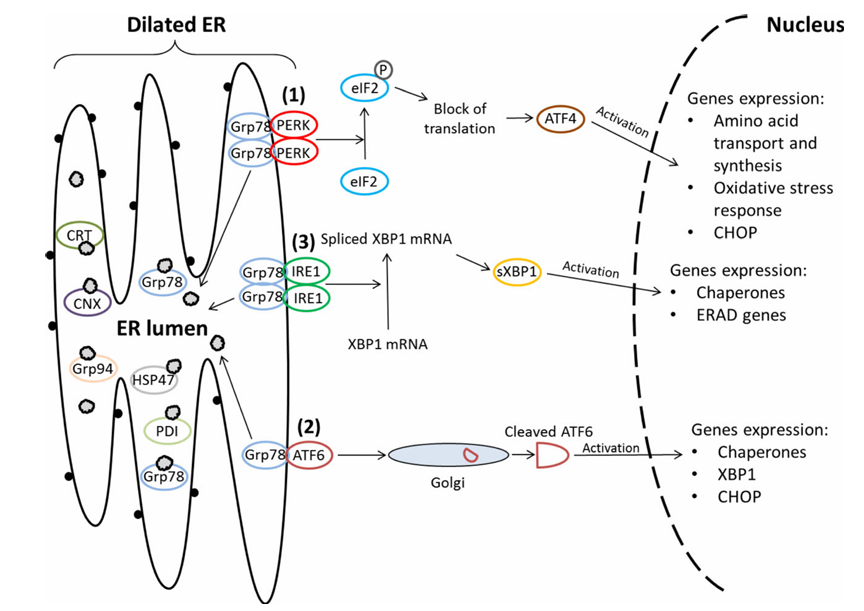
Figure 3. Schematic illustration of the three canonical branches of the unfolded protein response (UPR) activated during ER stress. Misfolded proteins activate PERK, ATF6, and IRE1, leading to downstream gene expression of ER chaperones, stress response factors, and apoptosis mediators. Adapted from Gawron K, 2016 [34].
Disruption of chondrogenic signaling pathways: These intracellular disruptions also intersect with pathways that regulate chondrocyte development. The provided sources document altered chondrocyte differentiation and growth-plate disorganization associated with COL2A1 (type II collagen) mutation–induced matrix abnormalities and ER stress, and they report downstream changes in growth-plate signaling that accompany these structural and cellular dysfunctions [34]. Gawron (2016) and the cited COL2A1 mouse-model studies describe intracellular retention of mutant type II procollagen, ER distension, and activation of the ER stress–UPR leading to apoptosis in affected chondrocytes [34]. These studies further report reduced extracellular type II collagen deposition, disrupted collagenous matrix architecture within the growth plate, and severe skeletal defects in homozygous mutant animals, indicating disturbed cartilage shaping and slowed endochondral ossification [34]. Proteoglycan loss, diminished type II collagen, and a reduction in SOX9 expression were documented in mutant homozygotes, demonstrating that key elements of the chondrogenic program are altered in the disease models [34]. Nevertheless, although associative changes in chondrogenic markers and growth-plate organization are described, the supplied reports do not provide direct mechanistic demonstrations that COL2A1 mutant proteins causally perturb canonical SOX9-dependent transcriptional control or the transforming growth factor β/bone morphogenetic protein (TGF β/BMP) signaling cascades [33,34]. Consequently, explicit claims that mutant type II collagen directly alters SOX9, TGF β, or BMP signaling exceed the direct evidence available in the provided sources and should be presented as hypotheses requiring further mechanistic investigation [34]. The interplay between BMP signaling, ER stress sensors, and chondrogenic transcription factors has been mapped in several experimental models, providing a broader context for how stress responses influence differentiation. As shown in Figure 4, bone morphogenetic protein 2 (BMP2) binds to its receptor and activates Smad1/5 and Smad4, which translocate to the nucleus and regulate the expression of key UPR-related genes such as XBP1 and ATF4, as well as the chondrogenic transcription factor SOX9. In parallel, BMP2 signaling enhances the activity of ER stress sensors PERK, ATF6, and IRE1, reinforcing the unfolded protein response and linking it directly to chondrocyte differentiation and cartilage development [42].
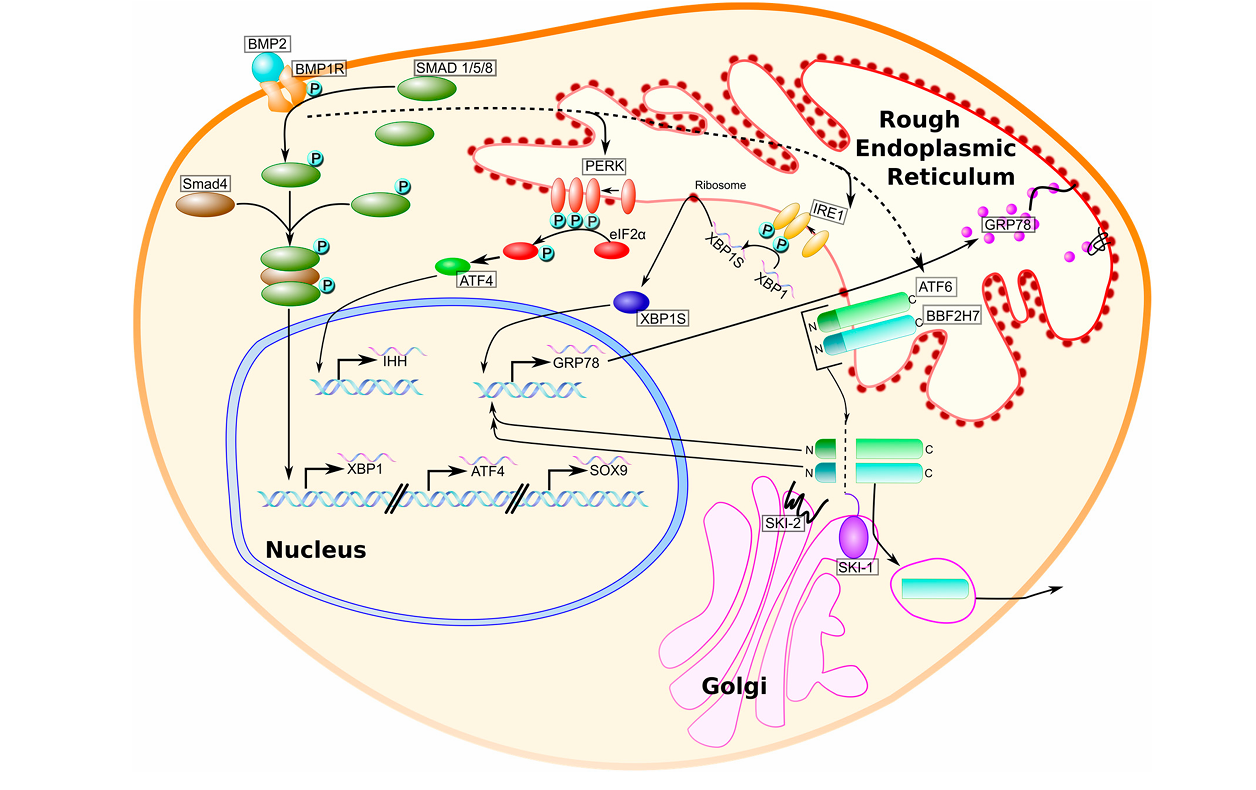
Figure 4. BMP2 activates UPR signaling pathways (PERK, ATF6, IRE1), which regulate SOX9, ATF4, and XBP1 to support chondrocyte differentiation and ER homeostasis (adapted from Hughes et al., 2017 [42]).
Associated skeletal dysplasias
Achondrogenesis Type II: Achondrogenesis type II is a rare, typically lethal skeletal dysplasia within the spectrum of type II collagenopathies [44]. Affected individuals characteristically show severe shortening of the limbs (micromelia), a markedly small thoracic cage, and a prominent or distended abdomen consistent with a short-limbed dwarfism phenotype [44,45]. The disorder results from dominant pathogenic variants in the COL2A1 gene (collagen, type II, alpha 1). A substantial proportion of disease-causing missense substitutions affect glycine residues within the Gly-X-Y repeat of the collagen triple helix. Such glycine substitutions produce dominant negative effects that interfere with homotrimer assembly and destabilize the triple helix, thereby generating the severe phenotypes observed in achondrogenesis type II and related disorders [44]. By contrast, truncating variants (for example, nonsense substitutions or out-of-frame deletions) commonly induce nonsense-mediated mRNA decay with resultant haploinsufficiency and are more frequently associated with milder clinical presentations [44]. Moreover, Bruni et al. (2021) note that the phenotypic consequence of in-frame deletions in COL2A1 appears to be influenced by both the size and the position of the deletion within the triple-helical region, with deletions of 18 amino acids or fewer often linked to less severe phenotypes and larger deletions tending to produce more disruptive structural defects and more severe disease [44].
Spondyloepiphyseal dysplasia congenita (SEDC): Spondyloepiphyseal dysplasia congenita (SEDC) is a recognized subtype within the spectrum of type II collagenopathies and is characterized at presentation by disproportionate short-trunk dwarfism with abnormal epiphyseal development and flattened vertebral bodies, often accompanied by odontoid hypoplasia [44]. Hip involvement in SEDC commonly manifests as structural deformities such as coxa vara and may progress to degenerative joint disease and early osteoarthritis [46]. At the molecular level, most SEDC cases result from missense variants in the collagen type II alpha-1 gene (COL2A1), with substitutions of glycine residues within the Gly-X-Y triple-helix motif being particularly frequent. These glycine replacements exert dominant-negative effects that disrupt triple-helix assembly [41,46]. Substitutions that replace glycine can destabilize the triple helical structure of type II collagen, impair intracellular processing and extracellular secretion, and thereby compromise collagen stability and function in cartilage [44]. Clinically, SEDC displays marked heterogeneity and may be accompanied by extraskeletal features, including ocular abnormalities (for example, myopia or vitreoretinal problems) and sensorineural hearing loss [46,47]. Further, identical COL2A1 mutations have been reported to produce variable phenotypes even among members of the same family, underscoring substantial intra- and interfamilial phenotypic variability and overlap across the type II collagenopathy spectrum [41,47,48].
Stickler syndrome: Stickler syndrome type I commonly presents with high myopia and carries a substantial risk of rhegmatogenous retinal detachment, with retinal detachment reported in 41.7% of cases in an East Asian systematic review [7]. Type II collagen (COL2A1)-related disease has a multisystem presentation that includes ocular, auditory, and musculoskeletal manifestations and may involve craniofacial abnormalities such as cleft palate. The clinical spectrum ranges from perinatally lethal disorders to milder, later-onset systemic features. The syndrome is typically inherited in an autosomal-dominant pattern [46]. In the East Asian review, truncating variants in the COL2A1 gene (nonsense and frameshift mutations) were identified and were associated with relatively milder systemic phenotypes compared with other variant classes. Conversely, comparative analyses indicated that splicing mutations are associated with more severe systemic manifestations than truncating mutations [7]. Phenotypic expression is highly variable among affected individuals, supporting a need for individualized clinical management and surveillance [7,46].
Genotype–phenotype correlation: Even when the same COL2A1 alteration is present, phenotypic expression can be highly variable between and within families, reflecting substantial intrafamilial heterogeneity [47]. Large systematic reviews and curated databases report hundreds of distinct COL2A1 variants (for example, 415 variants recorded across more than 700 patients), yet the clinical consequences of individual variants are not always predictable from mutation class alone [33,41]. Evidence from case series, functional assays, and genotype–phenotype analyses indicates that the precise amino acid substituted, the codon position within the alpha-1(II) chain, the size and location of in-frame deletions, and the expression level of the mutant product, together with other genetic or environmental modifiers, can modulate disease severity [41,44]. For these reasons, interpreting pathogenicity and prognosticating clinical outcome based solely on genotype remains problematic, which motivates the continued aggregation of high-quality phenotype–genotype data and functional validation studies [41]. A comparison of key clinical features, genetic mechanisms, inheritance patterns, and references associated with major skeletal dysplasias linked to COL2A1 mutations are summarized in Table 1.
|
Disorder |
Key Clinical Features |
Genetic Basis |
Inheritance Pattern |
Reference(s) |
|
Achondrogenesis Type II |
Severe micromelia, small thoracic cage, prominent abdomen, perinatal lethality |
Dominant COL2A1 mutations; mostly glycine substitutions in Gly-X-Y domain |
Autosomal dominant |
[44,45] |
|
Spondyloepiphyseal Dysplasia Congenita (SEDC) |
Short-trunk dwarfism, epiphyseal dysplasia, odontoid hypoplasia, hip deformity |
Missense COL2A1 mutations; frequent glycine substitutions in triple helix |
Autosomal dominant |
[47,48] |
|
Stickler Syndrome Type I |
High myopia, retinal detachment, hearing loss, joint hypermobility, cleft palate |
Truncating (nonsense/frameshift) and splice-site COL2A1 mutations |
Autosomal dominant |
[7,46] |
|
Genotype–Phenotype Correlation |
Phenotypic variability within and between families, unpredictable outcomes |
Variation in mutation type, codon position, deletion size, and expression level |
Varies; depends on mutation characteristics |
[33,44] |
Induced pluripotent stem cells (iPSCs)
Induced pluripotent stem cells (iPSCs) are artificially generated stem cells that originate from somatic cells reprogrammed through the co-expression of specific pluripotency-associated transcription factors [49]. Similar to embryonic stem cells, iPSCs possess the intrinsic ability to proliferate indefinitely under in vitro conditions and differentiate into derivatives of all three primary germ layers, ectoderm, mesoderm, and endoderm, as well as germ cells that form gametes [49]. Due to these defining properties, iPSCs serve as a versatile platform for biomedical research, including applications in drug discovery, disease modeling, toxicological studies, and regenerative medicine [49].
Discovery and reprogramming technologies
Yamanaka factors and alternatives:
Definition and discovery: The concept of reprogramming somatic cells into an induced pluripotent state was established in 2006, when Takahashi and Yamanaka demonstrated that a minimal set of transcription factors could reprogram differentiated mouse cells to pluripotency [50]. Specifically, Takahashi and Yamanaka screened candidate factors and identified four transcription factors—Oct4, Sox2, Klf4, and Myc—that were sufficient to induce pluripotency in mouse embryonic fibroblasts, creating the foundation for the OSKM paradigm [51]. Mechanistically, forced expression of these reprogramming factors drives coordinated changes in gene expression, and chromatin structure reprogramming entails erasure of somatic cell signatures, extensive chromatin remodeling, and activation of pluripotency-associated enhancers and genes [50]. The OSKM factors act in concert to displace somatic transcriptional regulators from somatic enhancers and to activate pluripotency enhancers, with reprogramming proceeding through early stochastic and later more deterministic transcriptional phases [50]. Although iPSCs share many features with embryonic stem cells and can be directed to differentiate into a wide range of human cell types, residual somatic epigenetic memory and reprogramming-associated heterogeneity can persist and influence the properties of derived iPSC lines [52].
Reprogramming limitations: Reprogramming somatic cells to iPSCs is limited by low efficiency during the early, largely stochastic phase of induction, the use of the proto-oncogene c-MYC—which increases reprogramming efficiency poses oncogenic safety concerns for clinical translation, and incomplete erasure of somatic epigenetic marks together with aberrant DNA methylation can persist in derived iPSC lines and alter their differentiation potential [50,52,53].
Alternative factor combinations: Alternative transcription factor combinations to the canonical OCT4, SOX2, KLF4, and c-MYC (OSKM) reprogramming set have been demonstrated in human cells; for example, human reprogramming protocols using OCT4 and SOX2 together with NANOG and LIN28 illustrate that distinct factor sets can induce pluripotency [50,51].
Non-integrative/zero-footprint delivery: Non-integrative and zero-footprint delivery approaches, such as Sendai virus, episomal plasmids, sequential mRNA transfection, and protein delivery, have been developed to avoid the stable genomic integration of exogenous reprogramming sequences, thereby improving the clinical suitability of generated iPSC lines [51,53].
Chemical reprogramming and small-molecule augmentation: Chemical reprogramming and small-molecule augmentation have been explored as defined, potentially scalable methods to induce pluripotency or to enhance reprogramming efficiency, and recent reports document progress toward chemical reprogramming of human cells; histone deacetylase inhibitors such as valproic acid are named as agents that can promote reprogramming [50,53].
Sources of somatic cells:
Fibroblasts: Skin fibroblasts were the first somatic cell type reprogrammed to induced pluripotent stem cells and remain the most commonly used primary cell source because they are inexpensive to maintain, have high viability and culture stability over multiple passages, and generally facilitate successful iPSC generation [52].
Peripheral blood mononuclear cells (PBMCs): Peripheral blood cells, including peripheral blood mononuclear cells (PBMCs), represent an accessible and less-invasive alternative to skin biopsy for obtaining donor material for reprogramming, although reprogramming efficiency and downstream differentiation behavior may differ from fibroblast-derived lines [52].
Keratinocytes: Keratinocytes obtained from plucked hair combine noninvasive collection, straightforward transport, and relatively high reprogramming efficiency, and are therefore recommended in many settings where noninvasive sampling is advantageous [52].
Urine-derived epithelial cells: Urine-derived somatic cells can be reprogrammed into urine-derived induced pluripotent stem cells (u-iPSCs), providing a noninvasive, readily obtainable source that has been differentiated into multiple clinically relevant cell types such as cardiomyocytes, neurons and astrocytes, hepatocyte-like cells, retinal cells, and kidney precursor cells, thus expanding options for patient-specific models and regenerative applications [54].
Considerations: The choice of somatic cell source involves practical trade-offs in collection methods and donor burden. Skin punch biopsies are invasive, whereas blood draws, hair plucks, and urine collections are less invasive. These factors influence feasibility in both clinical and research settings [52]. Biological trade-offs include differences in genomic integrity and residual somatic epigenetic memory: for example, skin fibroblasts may harbor ultraviolet-related DNA damage that increases mutational burden in derived iPSC lines, and persistent somatic transcriptional and DNA methylation signatures can bias differentiation outcomes, highlighting the need for standardized reprogramming protocols and quality control [50,53,54].
Advantages of patient-derived iPSCs
Disease modeling : To develop an in vitro disease model of human chondrodysplasia, investigators have used CRISPR/Cas9 gene editing to introduce a heterozygous COL2A1 exon 50 c.3508 GGT>TCA (p.G1170S) mutation into a control human induced pluripotent stem cell (iPSC) line, thereby generating defined mutant iPSC lines for mechanistic study [55]. In a related approach, a heterozygous COL2A1 p.G1113C mutation was introduced into an established control human iPSC line (MCRIi019-A) via CRISPR-Cas9, producing a gene-edited clone (MCRIi019-A-7) that retained normal iPSC morphology, expressed pluripotency markers, differentiated into the three embryonic germ layers, and maintained a normal karyotype, thereby establishing the edited lines as suitable substrates for downstream disease-relevant differentiation [56]. Using genetically defined wild-type and Arg719Cys COL2A1 iPSC lines, directed differentiation to chondrocytes produced cartilage tissues that were subjected to immunohistochemistry, electron microscopy, SDS-PAGE, and RNA-sequencing, enabling direct phenotypic comparison between isogenic genotypes [57]. Analyses of these iPSC-derived cartilage models revealed disease-relevant proteostasis phenotypes: Arg719Cys procollagen-II was modestly intracellularly retained and associated with endoplasmic reticulum (ER) storage defect and ER distention, while quantitative interactomic and transcriptional data demonstrated that the ER proteostasis network did not detectably engage the retained mutant procollagen-II [57].
Drug screening: Expandable mesenchyme-like derivatives of human induced pluripotent stem cells (ExpLBM), generated using limb-bud–directed protocols, have been induced and expanded for use in high-throughput drug screening, and ExpLBM differentiated from iPSCs of type II collagenopathy (COL2A1) patients were employed to develop a high-throughput screening system [58]. The induced pluripotent stem cell (iPSC)-derived cartilage model provides a genetically defined and expandable human-based system for dissecting mechanisms of failed proteostasis in collagenopathies, wherein malformed type II collagen (COL2A1) procollagen accumulates intracellularly and is secreted, contributing to deposition of a structurally compromised extracellular matrix [57]. Cord blood mononuclear cell–derived human iPSCs (CBMC-hiPSCs) undergo robust chondrogenic differentiation in pellet culture, producing cartilage-like pellets that exhibit high expression of aggrecan (ACAN), cartilage oligomeric matrix protein (COMP), type II collagen (COL2A1), and SOX9, and these pellets show higher collagen type II than collagen type I, supporting their potential utility for regenerative applications and as patient-relevant substrates for testing therapeutic candidates [59].
Genetic correction potential: CRISPR/Cas9 gene editing has been employed to introduce heterozygous type II collagen (COL2A1) mutations into control human induced pluripotent stem cell (iPSC) lines, generating isogenic mutant and parental control cell lines that serve as in vitro disease models of COL2A1-related chondrodysplasias [55,56]. Those genetically defined iPSC resources, when combined with directed differentiation into chondrocytes, constitute a human-based platform for investigating how specific COL2A1 genotypes affect cartilage phenotypes and for evaluating therapeutic strategies aimed at correcting pathologic procollagen folding and secretion [55,57]. Moreover, chondrogenic constructs derived from induced pluripotent stem cells can recapitulate cartilage-like features—cord blood mononuclear cell-derived hiPSCs (CBMC-hiPSCs) generate chondrogenic pellets with robust expression of cartilage markers such as ACAN, COMP, COL2A1, and SOX9—and CBMC-hiPSCs have been proposed as a potential clinically scalable cell source for cartilage regeneration [59].
Differentiation of induced pluripotent stem cells (iPSCs) into chondrocytes
Protocols and challenges: Multiple, well-established strategies are used to differentiate human iPSCs into chondrogenic lineages, including:
- Derivation of an intermediate mesenchymal-like population (induced mesenchymal stem cells; iMSCs) followed by high-density three-dimensional (3D) chondrogenic induction.
- Embryoid body (EB)–based differentiation with subsequent outgrowth and pellet formation.
- Directed, staged monolayer protocols that modulate developmental pathways.
- Co-culture with primary chondrocytes to provide paracrine guidance cues [60,61].
In the iMSC-intermediate approach, iPSCs are first converted into an expandable mesenchymal progenitor population (via direct plating or EB outgrowth), after which chondrogenic differentiation proceeds in 3D pellet or micromass culture with transforming growth factor-β (TGF-β) and bone morphogenetic protein (BMP) family stimulation to mimic pre-cartilage condensation and promote matrix production [62,63]. A validated Bio-protocol describes step-by-step iMSC derivation (direct plating and EB routes) and chondrogenic induction in 3D pellets or micromass, together with histological and quantitative reverse transcription polymerase chain reaction (qRT-PCR) readouts that detect temporal expression of cartilage genes and ECM accumulation in vitro [62]. EB-based, serum-/feeder-free protocols seeded 500 cells/well in ultra–low-attachment plates, transferred EBs onto Matrigel-coated plates in chondrogenic medium supplemented with TGF-β3, and subsequently re-aggregated cells into pellets of ~1.5×10^5 cells. 3D pellet culture promoted expression of chondrogenesis-related genes (FGFR3, SMAD3, NCAM1) and showed histological evidence of cartilage-like ECM with Alcian blue, Safranin O, and Toluidine blue staining, as well as lacuna formation [64]. Ekholm et al. (2025) describe a two-step chondrogenic protocol in which iPSCs undergo 14 days of monolayer induction with WNT3A, the SMAD/BMP inhibitor dorsomorphin, and fibroblast growth factor-10 (FGF10) to generate chondroprogenitors (iCHOp), followed by 3D free-floating pellet culture that further matures the cells, with the 3D medium containing TGF-β1 and BMP-2. Reproducibility and good manufacturing practice (GMP) compliance were tested by expanding iCHOp from three iPSC lines in a defined, xeno-free medium (PurStem 2), showing proliferative capacity and successful expansion in this serum-free medium. Evidence from iPSC-based chondrogenic protocols indicates that 3D formats, including high-density pellets and micromass, support chondrogenic differentiation, evidenced by elevated expression of chondrogenesis-related genes and deposition of proteoglycan-rich ECM. In serum- and feeder-free protocols, a 3D pellet phase yielded scaffoldless chondrogenic-like spheres with histochemical ECM signals and was described as mimicking avascular, diffusion-dependent aspects of cartilage. In iPSC-derived mesenchymal cells, 3D pellets exhibited significant increases in chondrogenic markers (e.g., COL2A1, COL9A1, COL11A1, SOX9, ACAN) along with Alcian blue–detectable glycosaminoglycans and collagen type II immunohistochemistry (IHC), while micromass cultures demonstrated proteoglycan accumulation [62–64]. Co-culture approaches, either direct or indirect, have also been used to bias iPSC differentiation toward chondrocytes, though they require access to primary chondrocytes and can suffer from potential contamination of co-cultured cell types [60].
Key challenges and translational considerations: First, safety concerns are highlighted. Csobonyeiova et al. (2021) reported the risk of teratoma formation, noting that one large tumor developed in a single mouse at 16 weeks, and emphasized that major attention must be focused on the purification of fully differentiated iPSC-derived chondrocytes before transplantation. By contrast, in subcutaneous transplantation experiments, no tumor formation was detected. Second, imperfect differentiation protocols still carry the risk of heterogeneity. The iCHOp exhibited a fetal-like chondrocyte phenotype characterized by versican expression, and residual epigenetic memory was identified as a significant factor affecting the standardization of iPSC lines [65,66]. Third, reliance on fetal bovine serum (FBS) and feeder layers introduces major obstacles. Batch-to-batch variation in FBS can alter differentiation efficiency and tissue quality, while animal-derived supplements raise infection risks and ethical concerns, ultimately disqualifying such products from clinical use. To overcome these limitations, Lach et al. (2019) developed a strictly defined and controllable protocol that eliminates serum and feeder cells, applying chemically defined conditions that may support clinical translation. They further emphasized the necessity of reproducible and standardized iPSC differentiation protocols. More broadly, Eremeev et al. (2023) highlighted that no generally accepted protocols have yet been approved for clinical trials, and that optimizing cultivation and differentiation conditions remains essential to enhance chondrogenesis. Finally, donor- and disease-specific variability further complicates outcomes. iPSCs derived from osteoarthritis (OA) cartilage (OA-iPSCs) show a significantly reduced capacity for chondrogenic differentiation, relatively smaller pellets, and lower expression of SOX9 and SOX9-regulated genes (COL2A1, ACAN) compared with healthy donor iPSCs. These findings highlight the impact of disease-specific epigenetic and metabolic memory and underscore the need for pathway-level rescue strategies [66,67].
iPSC models of chondrodysplasias
Existing models for skeletal diseases: iPSCs have emerged as a disease model for human chondrodysplasias: they can be generated from patient fibroblasts and carry disease-associated mutations, and iPSC-derived cartilage models can be used to investigate disease mechanisms and for drug screening and evaluation of therapeutic approaches [68,69]. Gain-of-function mutations in the fibroblast growth factor receptor 3 gene (FGFR3) result in skeletal dysplasias such as thanatophoric dysplasia type I (TD1) and achondroplasia (ACH) [69]. Patient fibroblasts from TD1 and ACH individuals can be converted into iPSCs and, after chondrogenic differentiation, the TD1- and ACH-iPSC-derived particles exhibit degraded cartilage with little Safranin O staining and decreased expression of cartilage matrix genes (COL2A1, ACAN) compared with wild-type iPSC-derived particles [69]. Chondrogenically differentiated FGFR3-mutant iPSCs show decreased proliferation and increased apoptosis, recapitulating primary abnormalities observed in FGFR3-related patients and models [69]. Importantly, statin treatment (for example, lovastatin or rosuvastatin) restored cartilage formation in TD1- and ACH-derived iPSC cartilage and increased bone lengths in an ACH mouse model. Mechanistically, statin exposure reduced the elevated FGFR3 protein and phosphorylated MAPK while increasing chondrogenic markers and proliferation, suggesting statin-induced degradation of mutant FGFR3 mainly through a proteasomal pathway [69].
Specific studies on COL2A1-related disorders: Type II collagen is the major structural protein of cartilage extracellular matrix (ECM), and dominant mutations in COL2A1 produce a broad spectrum of chondrodysplasias [55]. Several recent human iPSC resources have been produced by CRISPR/Cas9 editing of control iPSC lines to introduce heterozygous COL2A1 mutations. These edited lines and their isogenic controls exhibit canonical pluripotency marker expression and retain the capacity to differentiate into endodermal, ectodermal and mesodermal lineages, making them suitable platforms for cartilage disease modelling [55,68]. For example, the CRISPR-generated COL2A1 p.R989C line is reported to model a molecular defect previously linked to spondyloepiphyseal dysplasia congenita (SEDC), namely destabilization of the collagen triple helix with intracellular retention of misfolded collagen and potential activation of an unfolded protein response [68]. Likewise, a CRISPR-generated heterozygous COL2A1 p.G1170S iPSC line was produced to study type II collagenopathy mechanisms. Complementary work in a homozygous p.G1170S mouse reported endoplasmic reticulum (ER) accumulation of misfolded procollagen with activation of ER stress and apoptosis in growth-plate cartilage [55]. More recently, the Arg719Cys substitution in COL2A1 was studied using isogenic human iPSC lines, where mutant chondrocytes exhibited excessive post-translational modification of procollagen-II, partial intracellular retention with ER dilation, and deposition of a sparse, structurally deficient collagen-II matrix despite robust chondrogenic differentiation [35]. Because these COL2A1 models employ isogenic wild-type controls and CRISPR/Cas9 editing to introduce single substitutions, they permit direct linkage of genotype to phenotype, thereby providing mechanistic insight into type II collagenopathies [55,68,35]
Phenotypic and functional characterization
Lee et al. (2015) describe a growth factor–based differentiation protocol that yields articular-like hiPSC-derived chondrocytes in approximately two weeks at >90% efficiency, and they performed molecular characterization to define the differentiation program and cell identity. The hiChondrocytes produced abundant ACAN and COL2A1, retained SOX9/COL2 expression after passaging, and formed cartilage in vivo without tumor formation, supporting their suitability for regenerative and disease-modeling applications [70].
Morphological assessment: Building on these findings, efficient hiPSC chondrogenic protocols generate 3D pellets, spheroids, or sheet-like chondrocyte tissues that are analyzed by histology and used for in vivo engraftment studies. These constructs commonly produce cartilage-associated matrix components including GAGs, COL2, and ACAN [70–72]. In this context, both hiPSC-derived pellet cultures and hiPSC expandable limb-bud mesenchymal (ExpLBM) chondrocyte sheets show positive histochemical staining for proteoglycans/glycosaminoglycans. Safranin-O staining demonstrated production of glycosaminoglycans in hiPSC-derived chondrocyte pellet cultures, and ExpLBM-derived chondrocyte sheets were embedded in extracellular matrix that stained intensely and homogeneously with Alcian blue and Safranin O, consistent with hyaline cartilage–like extracellular matrix accumulation [70,71]. Moreover, Xiong et al. (2025) reported that immunostaining and histological staining (including COL2 and Safranin O) revealed cartilaginous matrix deposition, and that engineered osteochondral fusion constructs formed polarized, growth plate-like structures characterized by columnar proliferative chondrocytes arranged between resting and hypertrophic zones [72].
Molecular characterization: Transitioning from morphology to molecular profiling, both Lee et al. (2015) and Takao et al. (2023) report that gene-level analyses detect chondrogenic transcription factor expression (SOX9) alongside elevated expression of cartilage matrix genes, including COL2A1 and ACAN, in hiPSC-derived chondrocyte preparations [70,71]. In addition, hiPSC-derived articular-like chondrocytes express high COL2A1 and ACAN but show little staining or gene expression for the hypertrophy marker COL10A1, consistent with an articular phenotype. A developmentally directed scl-progenitor strategy can lead engineered cartilage to progress through hypertrophy to vascular invasion and bone formation, representing endochondral ossification [70,72]. Furthermore, genome-scale transcriptional profiling combined with single-cell flow cytometry has been used to track SOX9 alongside the surface markers CD44 and CD140, revealing distinct expression states that correlated with SOX9 dynamics during differentiation. Nearly all terminal hiChondrocytes acquired a SOX9-high, CD140-high, CD44-high phenotype [70].
Biochemical and functional assays: Moving beyond gene expression, hiPSC-derived chondrocytes produce cartilaginous matrix, as evidenced by Safranin-O staining for glycosaminoglycans and abundant aggrecan (ACAN) and type II collagen in pellet cultures, with quantitative assays showing greater total GAG accumulation in hiChondrocyte pellets compared to adult chondrocyte pellets [70]. In line with these biochemical findings, iPSC extracellular vesicles (iPSC-EVs) enhance primary human chondrocyte proliferation and suppress senescence through regulation of cyclin-dependent kinase inhibitor 1 (p21) and type II collagen. They reduce matrix-degrading enzymes and interleukin-6 (IL-6) while attenuating chondrocyte cell death, and influence the macrophage milieu, thereby indirectly ameliorating chondrocyte degradation in vivo. Anterior cruciate ligament transection (ACLT) joints treated with iPSC-EVs exhibited stronger collagen II and lower MMP13, a disintegrin and metalloproteinase with thrombospondin motifs 5 (ADAMTS5), and tumor necrosis factor alpha (TNFα) expression compared with untreated ACLT controls [73]. Complementing these biochemical insights, functional testing ranges from biological functional assays to biomechanical evaluation. Engineered constructs derived from hPSC progenitors have been probed mechanically (for example by nanoindentation to determine region-specific Young’s modulus), and in vivo transplantation studies test engraftment, matrix deposition and phenotypic stability in repair or ossification paradigms [71,72].
Consistently, iPSC-derived extracellular vesicles promoted chondrocyte proliferation and suppressed senescence via regulation of p21 and collagen II, reduced IL-1β–driven MMP13 and IL-6 expression, and decreased IL-1β–induced chondrocyte death in vitro and after intra-articular injection in an ACLT rabbit model. These effects were associated with increased collagen II and decreased MMP13, ADAMTS5, and TNF-α in cartilage. Modulation of macrophage polarization and cytokine secretion was reported as a likely intermediary mechanism [73]. Taken together, the published protocols and validation studies illustrate that a combined approach morphological assessment (3D constructs, histology, microscopy), molecular profiling (SOX9, COL2A1, ACAN and hypertrophy markers), biochemical/protein assays (GAG quantification, western blot/ELISA, MMP arrays), and functional testing (mechanics, engraftment, EV-mediated modulation)—provides a comprehensive framework for confirming the phenotype and functional competence of iPSC-derived chondrocytes for both modeling and regenerative applications [70–,73].
CRISPR-Cas9 and Gene editing in iPSCs
The CRISPR-Cas9 system enables targeted genome editing in iPSCs by using a guide RNA to direct Cas9 to specific genomic loci, creating double-strand breaks repaired by non-homologous end joining (NHEJ) or homology-directed repair (HDR) [3,74,75]. This approach allows the generation of isogenic lines differing only at a disease-causing locus, facilitating mechanistic studies of disease phenotypes and testing of therapeutic correction strategies. Engineered Cas9 variants and base editors have further improved editing precision and minimized off-target effects [3]. In COL2A1 studies, researchers introduced heterozygous COL2A1 mutations into control iPSCs to model type II collagenopathies and demonstrated that these lines retained pluripotency and trilineage differentiation capacity [55, 68]. Reporter knock-in of COL2A1 (e.g., COL2A1-GFP) was further used to identify chondroprogenitor subpopulations, where cells sorted as platelet-derived growth factor receptor beta (PDGFRβ), melanoma cell adhesion molecule (CD146/MCAM), and activated leukocyte cell adhesion molecule (CD166/ALCAM) positive and CD45 negative produced more homogeneous cartilage-like matrix and upregulated chondrogenic markers (SOX9, COL2A1, ACAN) as illustrated in Figure 5 [76]. Collectively, these genome-editing approaches highlight the potential of CRISPR/Cas9-integrated iPSC models as a robust platform for dissecting COL2A1-associated mechanisms and developing targeted repair strategies.
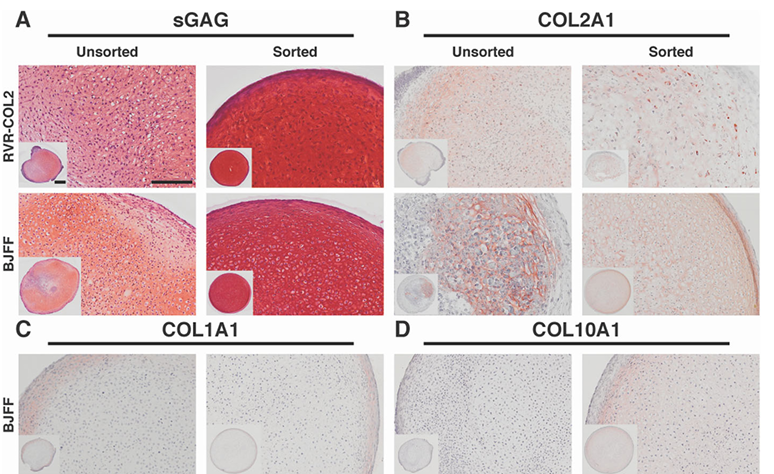
Figure 5. Increased sGAG/DNA content and upregulation of chondrogenic genes (SOX9, COL2A1, ACAN) in FACS-sorted PDGFRβ+/CD146+/CD166+/CD45− chondroprogenitors compared with unsorted populations. Adapted from Dicks et al. (2020) [76].
Challenges and limitations in modeling COL2A1 mutations
Although iPSC-based platforms for COL2A1-related cartilage disorders have substantially advanced mechanistic and translational studies, important technical, biological, and ethical barriers remain that constrain their immediate clinical applicability [77].
Technical hurdles in iPSC reprogramming and differentiation: Technical challenges include variability in reprogramming and differentiation: the donor cell type used to generate iPSCs is a significant source of variation, and differences in laboratory protocols and maturation stages between studies create inter-experimental heterogeneity that complicates comparisons [77]. Moreover, genetic and epigenetic differences, including culture-associated epigenomic or genomic alterations, can produce line-to-line variability and affect growth and differentiation behaviors, complicating interpretation of genotype–phenotype relationships [49,78]. In addition, current directed protocols frequently yield cells that are incompletely characterized and fail to recapitulate the transcriptomic profile of native cartilage, so differentiation protocols require further optimization before reliably producing mature articular chondrocytes [77]. Incomplete maturation is common with currently published protocols, and because these cells do not reproduce the native cartilage transcriptome, protocol optimization is needed before reliably generating mature articular chondrocytes for broad application [77]. Finally, reproducibility is limited: different laboratories use different differentiation protocols and assess cells at varying maturation stages, and differentiation variability is a common concern even for the same iPSC line, complicating cross study comparisons [77].
Disease complexity and in vitro limitations: Beyond technical hurdles, additional challenges arise from disease complexity and the inherent limitations of in vitro models. Standard 2D in vitro formats lack three-dimensional tissue architecture, multicompartment integration, and the biomechanical loading of native cartilage and joints; organ-on-a-chip microphysiological systems are designed to address these shortcomings by recapitulating 3D organization, microenvironmental cues, and in vivo-like mechanical stimuli [78,79].
Furthermore, advanced microphysiological systems recreate three-dimensional tissue organization and microenvironmental cues to address limitations of conventional 2D cultures and to permit modeling of tissue interfaces and physiological loading, but the inherent complexity and load-bearing properties of musculoskeletal tissues introduce significant technical challenges that must be addressed [78].
In addition, iPSC-derived chondrocyte constructs frequently fail to reproduce the full transcriptomic profile of native cartilage and show only limited similarity to primary articular chondrocytes; longer in vitro maturation improves chondrogenic maturation, and modeling efforts should replicate the joint environment (including degenerative and mechanical stressors) when aiming to generate osteoarthritis-relevant models [77,78].
Ethical considerations in iPSC research: Finally, ethical and safety issues present additional barriers for clinical translation.
iPSC-derived models reproduce intracellular pathogenic features such as impaired matrix formation and intracellular aggrecan accumulation, and they have shown accumulation of improperly folded proteins in the endoplasmic reticulum that is associated with ER stress responses and chondrocyte apoptosis, which require careful molecular assays and longer observation windows to detect reliably [61,77]. Moreover, iPSC-derived constructs can produce hyaline cartilage and extracellular matrix in vivo, but at least one study reported a tumor developing after transplantation and recommended strict purification of fully differentiated iPSC-derived chondrocytes before clinical translation [61]. In addition to biological risks, ethical, legal, and safety considerations restrict iPSC research and clinical translation: informed voluntary consent is required for use of human cells, specialist guidelines (for example, International Society for Stem Cell Research [ISSCR]) and risk-based regulatory frameworks (including Good Manufacturing Practice [GMP] for Advanced Therapy Medicinal Products [ATMPs]) govern therapeutic induced pluripotent stem cell (iPSC) products, and genetic manipulations used to produce isogenic models carry safety risks that demand additional quality controls and clinical-grade manufacturing capacity [49].
Broader challenges and limitations of iPSC-based cartilage modeling: Variability in the behavior of iPSCs remains a critical obstacle to reliable modeling of cartilage. Various reports have identified that large differences exist between iPSC lines in their reprogramming efficiency and chondrogenic capacity due to residual epigenetic marks inherited from donor cells, which have been shown to influence lineage commitment [65,77,80]. These differences often manifest as immature, hypertrophic, or fibrotic extracellular matrices, lowering functional cartilage quality [80]. Further, even within one iPSC line, there is great fluctuation in experimental results, making it hard to reproduce between different laboratories [77]. Somatic origin has a significant impact on the differentiation of reprogrammed cells. iPSCs derived from fibroblasts, blood, or keratinocytes differ in chondrogenic potential due to epigenetic memory from their starting tissue [65,77]. Moreover, the iPSCs derived from chondrocytes of osteoarthritis retain disease-specific metabolic and epigenetic profiles, thereby limiting their chondrogenic differentiation capability; thus, pathological memory is also preserved after reprogramming [65].
However, although many differentiation protocols start with the formation of embryoid bodies and mesodermal induction, most of the research still reports immature chondrocytes being generated, depositing limited extracellular matrix, with hypertrophic tendencies [80–82]. Comparisons between different laboratories are difficult because there is no consistency in media, formulation of growth factors, or duration of maturation [77]. More importantly, most of the currently available in vitro systems fail to recapitulate the depth-dependent structural zonation and mechanical cues that are typical for native cartilage, and evidence exists that dynamic mechanical stimulation considerably enhances chondrogenic maturation in iPSC-based systems [80]. Organoid and bioprinted three-dimensional culture formats support chondrogenic gene expression and matrix formation to a significantly greater degree when compared to two-dimensional systems. However, even three-dimensional models fail to accurately recapitulate biomechanical and inflammatory interactions that define the in vivo joint environment [82]. This issue is very significant in the context of modeling osteoarthritis because iPSC-derived cells are very sensitive to inflammatory mediators such as IL-1β and TNF-α, whereas OA-derived iMSCs show increased inflammatory reactivity, underscoring the need to include synovial-like inflammatory cues for more relevant disease models [65]. Modeling of pathogenic mutations, including those in COL2A1 genes, through the use of iPSC-derived chondrocytes triggers the accumulation of misfolded collagen and subsequent endoplasmic-reticulum stress [77]. Moreover, the presence of residual undifferentiated iPSCs can lead to teratoma formation, while reprogramming by viral vectors increases the risk of genomic instability [81]. Clinical translation is further restricted by ethical, regulatory, and Good Manufacturing Practice considerations [81]. Characterization of iPSC-derived chondrocytes also remains a challenge; distinguishing mature articular chondrocytes from transient or hypertrophic phenotypes requires extensive marker panels and comparison with their in vivo expression profiles [77]. Finally, heterogeneity within the differentiated cultures persists, yielding mixed populations of variable maturity [77,80]. Continuous optimization of reprogramming and differentiation strategies, combined with automation and standardization, is still imperative to achieve functionally mature and clinically safe iPSC-derived chondrocytes [81,82].
Discussion / Future Perspectives
iPSC-based CRISPR/Cas9 genome editing has been used to generate heterozygous COL2A1 mutant human iPSC lines from established control lines, establishing in vitro disease models of type II collagenopathies [68].
Correction of patient mutations and creation of isogenic lines
In this context, researchers used CRISPR/Cas9 to introduce specific heterozygous COL2A1 point mutations (c.2965 C>T, p.R989C; c.3508 GGT>TCA, p.G1170S) into control human iPSC lines, and each edited line together with its parental isogenic control serves as an in vitro model for studying the mechanisms of type II collagenopathies [55,68]. Furthermore, gene-edited COL2A1 iPSC lines display typical pluripotent stem cell characteristics, including compact colony morphology and canonical pluripotency marker expression, and retain the capacity to differentiate into derivatives of all three embryonic germ layers, supporting their suitability for downstream cartilage differentiation experiments [55,68]. In addition, isogenic mutant control iPSC pairs have been presented as in vitro disease models to link COL2A1 genotypes with cellular phenotypes and to evaluate candidate therapeutic strategies for type II collagenopathies [55,68].
Elucidation of pathogenic pathways
Moving from correction studies to mechanistic insights, intracellular retention of misfolded type II procollagen, destabilization of the collagen triple helix, and initiation of an unfolded-protein response have been reported in human iPSC-derived lines carrying a heterozygous COL2A1 p.R989C mutation [68]. In addition, enriching PDGFRβ+/CD146+/CD166+/CD45− chondroprogenitors before directed chondrogenic differentiation markedly increases matrix production (sGAG/DNA ≈ 20 ng/ng) and strongly upregulates chondrogenic genes such as SOX9, COL2A1, and ACAN relative to unsorted populations as illustrated in Figure 5 [76].
Functional validation using reporter systems
Finally, reporter knock-in of COL2A1 (e.g., COL2A1-GFP) has been used to identify cell surface markers and prospectively isolate chondroprogenitors. Cells sorted by FACS as platelet-derived growth factor receptor beta (PDGFRβ), melanoma cell adhesion molecule (CD146/MCAM), and activated leukocyte cell adhesion molecule (CD166/ALCAM) positive and CD45 negative produced more homogeneous cartilage-like matrix, showed a large increase in sGAG/DNA (to ≈19.9–20 ng/ng), and exhibited markedly higher expression of chondrogenic genes (SOX9, COL2A1, ACAN) in pellet assays, as illustrated in Figure 5 [76].
Conclusion
Mutations in the COL2A1 gene result in defective collagen fibrillogenesis, disturbed extracellular matrix organization, and abnormal chondrocyte differentiation that together underline the skeletal phenotypes observed in type II collagenopathies. The molecular defects, such as glycine or arginine substitutions within the triple-helical region and mutations in the C-propeptide, lead to intracellular retention of misfolded procollagen, activation of endoplasmic reticulum stress responses, and impaired growth-plate organization. Patient-derived iPSCs carrying COL2A1 mutations reproduce these cellular alterations in vitro and thus provide a reliable model to study disease mechanisms at the molecular level. Moreover, CRISPR/Cas9-mediated editing enables the generation of isogenic control lines for direct phenotype comparison, the correction of pathogenic alleles, and the assessment of therapeutic strategies targeting COL2A1 variants. Collectively, these integrated approaches link genotype to cellular phenotype and represent a foundation for future translational applications in cartilage-related skeletal dysplasias.
Declarations
Funding
This research was supported by institutional funding.
Conflicts of interest / Competing interests
The authors declare that there are no conflicts of interest or competing interests related to this work.
Ethics approval
Not applicable. This review did not involve human participants, animal experiments, or clinical data.
Consent to participate
Not applicable.
Consent for publication
All authors have reviewed and approved the final version of the manuscript for publication.
Clinical trial number
Not applicable.
Availability of data and material
Not applicable. This is a literature-based review; no new datasets were generated or analyzed.
Code availability
Not applicable.
Authors’ contributions
Latifa Mohammed Aljuid conceptualized the study, performed the literature search, analyzed the data, and wrote the manuscript. Dr. Dareen Al-Yousfi, Dr. Manal Mosa Housawi, and Dr. Ayman Z. Elsamanoudy supervised the project, provided scientific guidance, and contributed to manuscript review and revision. All authors reviewed and approved the final version of the manuscript.
References
2. Sophia Fox AJ, Bedi A, Rodeo SA. The basic science of articular cartilage: structure, composition, and function. Sports Health. 2009 Nov;1(6):461–8.
3. Walsh C, Jin S. Induced Pluripotent Stem Cells and CRISPR-Cas9 Innovations for Treating Alpha-1 Antitrypsin Deficiency and Glycogen Storage Diseases. Cells. 2024 Jun 18;13(12):1052.
4. National Center for Biotechnology Information (NCBI). COL2A1 gene (Gene ID: 1280) [Internet]. Bethesda (MD): National Library of Medicine (US); 2024 [cited 2026 Jan 28]. Available from: https://www.ncbi.nlm.nih.gov/gene/1280
5. Deng H, Huang X, Yuan L. Molecular genetics of the COL2A1-related disorders. Mutat Res Rev Mutat Res. 2016 Apr-Jun;768:1–13.
6. Yeter B, Kendir Demirkol Y, Eser M, Akgülle AH, Sözeri B, Kırmızıbekmez H. Diagnostic Challenge of Phenotypic Variability in COL2A1-related Disorders: Four Novel Variants That Expand the Clinical Spectrum. J Clin Res Pediatr Endocrinol. 2025 Aug 22;17(3):297–306
7. Wang DD, Gao FJ, Hu FY, Zhang SH, Xu P, Wu JH. Mutation Spectrum of Stickler Syndrome Type I and Genotype-phenotype Analysis in East Asian Population: a systematic review. BMC Med Genet. 2020 Feb 10;21(1):27.
8. Markova T, Kenis V, Melchenko E, Osipova D, Nagornova T, Orlova A, et al. Clinical and Genetic Characteristics of COL2A1-Associated Skeletal Dysplasias in 60 Russian Patients: Part I. Genes (Basel). 2022 Jan 13;13(1):137.
9. Gong Y, Zhu W, Zhu M, Chen D, Wu S, Hu S, et al. Identification and functional characteristics of a novel splicing heterozygote variant of COL2A1 associated with Stickler syndrome type I. Front Genet. 2024 Jul 10;15:1308737.
10. Yasuda H, Oh CD, Chen D, de Crombrugghe B, Kim JH. A Novel Regulatory Mechanism of Type II Collagen Expression via a SOX9-dependent Enhancer in Intron 6. J Biol Chem. 2017 Jan 13;292(2):528–38.
11. Darbellay F, Ramisch A, Lopez-Delisle L, Kosicki M, Rauseo A, Jouini Z, et al. Pre-hypertrophic chondrogenic enhancer landscape of limb and axial skeleton development. Nat Commun. 2024 Jun 6;15(1):4820.
12. Smeriglio P, Grandi FC, Taylor SEB, Zalc A, Bhutani N. TET1 Directs Chondrogenic Differentiation by Regulating SOX9 Dependent Activation of Col2a1 and Acan In Vitro. JBMR Plus. 2020 Jun 26;4(8):e10383.
13. Zhou J, Yuan T. Pathogenicity effects of a COL2A1 missense mutation (c.1594G>C) in cartilage development. Transl Pediatr. 2025 Jul 31;14(7):1511–9.
14. Brachvogel B, Zaucke F, Dave K, Norris EL, Stermann J, Dayakli M, et al. Comparative proteomic analysis of normal and collagen IX null mouse cartilage reveals altered extracellular matrix composition and novel components of the collagen IX interactome. J Biol Chem. 2013 May 10;288(19):13481–92.
15. Zhang J, Zhang W, Shi J, Dai J, Shen SG. Dlx2 overexpression enhanced accumulation of type II collagen and aggrecan by inhibiting MMP13 expression in mice chondrocytes. Biochem Biophys Res Commun. 2018 Sep 5;503(2):528–35.
16. Reeck JC. The Role of Col11a1 Expression During Cartilage Development. Doctoral dissertation, University of South Carolina, 2017.
17. Oppenheimer H, Meir H, Zini A, Haze A, Kandel L, Mattan Y, et al. Set7/9 represses SirT1 to induce collagen type II expression in 3D cultured human chodrocytes. Osteoarthritis and Cartilage. 2013 Apr 1;21:S170.
18. Gabler J, Ruetze M, Kynast KL, Grossner T, Diederichs S, Richter W. Stage-Specific miRs in Chondrocyte Maturation: Differentiation-Dependent and Hypertrophy-Related miR Clusters and the miR-181 Family. Tissue Eng Part A. 2015 Dec;21(23-24):2840–51.
19. Lui JC, Yue S, Lee A, Kikani B, Temnycky A, Barnes KM, et al. Persistent Sox9 expression in hypertrophic chondrocytes suppresses transdifferentiation into osteoblasts. Bone. 2019 Aug;125:169–77.
20. Ono K, Hata K, Nakamura E, Ishihara S, Kobayashi S, Nakanishi M, et al. Dmrt2 promotes transition of endochondral bone formation by linking Sox9 and Runx2. Commun Biol. 2021 Mar 11;4(1):326.
21. Javaheri B, Caetano-Silva SP, Kanakis I, Bou-Gharios G, Pitsillides AA. The Chondro-Osseous Continuum: Is It Possible to Unlock the Potential Assigned Within? Front Bioeng Biotechnol. 2018 Mar 21;6:28.
22. Chen H, Tan X-N, Hu S, Liu R-Q, Peng L-H, Li Y-M, et al. Molecular Mechanisms of Chondrocyte Proliferation and Differentiation. Frontiers in Cell and Developmental Biology. 2021;9:664168.
23. Guasto A, Cormier-Daire V. Signaling Pathways in Bone Development and Their Related Skeletal Dysplasia. Int J Mol Sci. 2021 Apr 21;22(9):4321.
24. Li J, Dong S. The Signaling Pathways Involved in Chondrocyte Differentiation and Hypertrophic Differentiation. Stem Cells Int. 2016;2016:2470351.
25. Liu CF, Lefebvre V. The transcription factors SOX9 and SOX5/SOX6 cooperate genome-wide through super-enhancers to drive chondrogenesis. Nucleic Acids Res. 2015 Sep 30;43(17):8183–203.
26. Dexheimer V, Gabler J, Bomans K, Sims T, Omlor G, Richter W. Differential expression of TGF-β superfamily members and role of Smad1/5/9-signalling in chondral versus endochondral chondrocyte differentiation. Sci Rep. 2016 Nov 16;6:36655.
27. Dreher SI, Fischer J, Walker T, Diederichs S, Richter W. Significance of MEF2C and RUNX3 Regulation for Endochondral Differentiation of Human Mesenchymal Progenitor Cells. Front Cell Dev Biol. 2020 Mar 4;8:81.
28. Ashraf S, Cha BH, Kim JS, Ahn J, Han I, Park H, Lee SH. Regulation of senescence associated signaling mechanisms in chondrocytes for cartilage tissue regeneration. Osteoarthritis Cartilage. 2016 Feb;24(2):196–205
29. Fu B, Shen J, Zou X, Sun N, Zhang Z, Liu Z, et al. Matrix stiffening promotes chondrocyte senescence and the osteoarthritis development through downregulating HDAC3. Bone Res. 2024 May 24;12(1):32.
30. Cai W, Zhang Y, Jin W, Wei S, Chen J, Zhong C, et al. Procyanidin B2 ameliorates the progression of osteoarthritis: An in vitro and in vivo study. Int Immunopharmacol. 2022 Dec;113(Pt A):109336.
31. Xu F, Zhao LJ, Liao T, Li ZC, Wang LL, Lin PY, et al. Ononin ameliorates inflammation and cartilage degradation in rat chondrocytes with IL-1β-induced osteoarthritis by downregulating the MAPK and NF-κB pathways. BMC Complement Med Ther. 2022 Jan 27;22(1):25.
32. Sarkar A, Liu NQ, Magallanes J, Tassey J, Lee S, Shkhyan R, et al. STAT3 promotes a youthful epigenetic state in articular chondrocytes. Aging Cell. 2023 Feb;22(2):e13773.
33. Nenna R, Turchetti A, Mastrogiorgio G, Midulla F. COL2A1 Gene Mutations: Mechanisms of Spondyloepiphyseal Dysplasia Congenita. Appl Clin Genet. 2019 Dec 4;12:235–8.
34. Gawron K. Endoplasmic reticulum stress in chondrodysplasias caused by mutations in collagen types II and X. Cell Stress Chaperones. 2016 Nov;21(6):943–58.
35. Yammine KM, Mirda Abularach S, Xiong M, Kim SY, Bikovtseva AA, Butty VL, et al. Failed cellular surveillance enables pathogenic matrix deposition in a COL2A1-related osteoarthritis. J Biol Chem. 2025 Aug;301(8):110436.
36. Li LJ, Lyu F, Song YW, Wang O, Jiang Y, Xia WB, et al. Genotype-phenotype relationship in a large cohort of osteogenesis imperfecta patients with COL1A1 mutations revealed by a new scoring system. Chin Med J (Engl). 2019 Jan 20;132(2):145–53.
37. Lin HY, Chuang CK, Su YN, Chen MR, Chiu HC, Niu DM, et al. Genotype and phenotype analysis of Taiwanese patients with osteogenesis imperfecta. Orphanet J Rare Dis. 2015 Dec 1;10:152.
38. Zhang H, Yue H, Wang C, Hu W, Gu J, He J, et al. Clinical characteristics and the identification of novel mutations of COL1A1 and COL1A2 in 61 Chinese patients with osteogenesis imperfecta. Mol Med Rep. 2016 Nov;14(5):4918–26.
39. Gong Z, Wang H, Lin Z. Glycine substitution mutation of COL5A1 in classic Ehlers-Danlos syndrome: a case report and literature review. Clin Exp Dermatol. 2021 Jul;46(5):987–9.
40. Veres K, Nagy B, Ember Z, Bene J, Hadzsiev K, Medvecz M, et al . Increased Phenotype Severity Associated with Splice-Site Variants in a Hungarian Pediatric Neurofibromatosis 1 Cohort: A Retrospective Study. Biomedicines. 2025 Jan 9;13(1):146.
41. Barat-Houari M, Sarrabay G, Gatinois V, Fabre A, Dumont B, Genevieve D, et al. Mutation Update for COL2A1 Gene Variants Associated with Type II Collagenopathies. Hum Mutat. 2016 Jan;37(1):7–15.
42. Hughes A, Oxford AE, Tawara K, Jorcyk CL, Oxford JT. Endoplasmic Reticulum Stress and Unfolded Protein Response in Cartilage Pathophysiology; Contributing Factors to Apoptosis and Osteoarthritis. Int J Mol Sci. 2017 Mar 20;18(3):665.
43. Kimura M, Ichimura S, Sasaki K, Masuya H, Suzuki T, Wakana S, et al. Endoplasmic reticulum stress-mediated apoptosis contributes to a skeletal dysplasia resembling platyspondylic lethal skeletal dysplasia, Torrance type, in a novel Col2a1 mutant mouse line. Biochem Biophys Res Commun. 2015 Dec 4-11;468(1-2):86–91.
44. Bruni V, Spoleti CB, La Barbera A, Dattilo V, Colao E, Votino C, et al. A Novel Splicing Variant of COL2A1 in a Fetus with Achondrogenesis Type II: Interpretation of Pathogenicity of In-Frame Deletions. Genes (Basel). 2021 Sep 10;12(9):1395.
45. Maheshwari S, Ingole D, Chatterjee S, Rajesh U, Anand V. A case report of achondrogenesis type II (Langer-Saldino achondrogenesis). Egyptian Journal of Radiology and Nuclear Medicine. 2021 Apr 22;52(1):113.
46. Xu Y, Li L, Wang C, Yue H, Zhang H, Gu J, et al. Clinical and Molecular Characterization and Discovery of Novel Genetic Mutations of Chinese Patients with COL2A1-related Dysplasia. Int J Biol Sci. 2020 Jan 16;16(5):859–68.
47. Nakashima Y, Sakamoto Y, Nishimura G, Ikegawa S, Iwamoto Y. A novel type II collagen gene mutation in a family with spondyloepiphyseal dysplasia and extensive intrafamilial phenotypic diversity. Hum Genome Var. 2016 May 19;3:16007.
48. Huang X, Deng X, Xu H, Wu S, Yuan L, Yang Z, et al. Identification of a Novel Mutation in the COL2A1 Gene in a Chinese Family with Spondyloepiphyseal Dysplasia Congenita. PLoS One. 2015 Jun 1;10(6):e0127529.
49. Moradi S, Mahdizadeh H, Šarić T, Kim J, Harati J, Shahsavarani H, et al. Research and therapy with induced pluripotent stem cells (iPSCs): social, legal, and ethical considerations. Stem Cell Res Ther. 2019 Nov 21;10(1):341.
50. Cerneckis J, Cai H, Shi Y. Induced pluripotent stem cells (iPSCs): molecular mechanisms of induction and applications. Signal Transduct Target Ther. 2024 Apr 26;9(1):112.
51. Van Pham P. Direct reprogramming of somatic cells: an update. Biomedical Research and Therapy. 2015 Mar 13;2(3):8.
52. Raab S, Klingenstein M, Liebau S, Linta L. A Comparative View on Human Somatic Cell Sources for iPSC Generation. Stem Cells Int. 2014;2014:768391.
53. Nath SC, Menendez L, Friedrich Ben-Nun I. Overcoming the Variability of iPSCs in the Manufacturing of Cell-Based Therapies. Int J Mol Sci. 2023 Nov 29;24(23):16929.
54. Yin X, Li Q, Shu Y, Wang H, Thomas B, Maxwell JT, et al. Exploiting urine-derived induced pluripotent stem cells for advancing precision medicine in cell therapy, disease modeling, and drug testing. J Biomed Sci. 2024 May 9;31(1):47.
55. Kung LHW, Sampurno L, Yammine KM, Graham A, McDonald P, Bateman JF, et al. CRISPR/Cas9 editing to generate a heterozygous COL2A1 p.G1170S human chondrodysplasia iPSC line, MCRIi019-A-2, in a control iPSC line, MCRIi019-A. Stem Cell Res. 2020 Oct;48:101962.
56. Lilianty J, Bateman JF, Lamandé SR. Generation of a heterozygous COL2A1 (p.G1113C) hypochondrogenesis mutation iPSC line, MCRIi019-A-7, using CRISPR/Cas9 gene editing. Stem Cell Res. 2021 Oct;56:102515.
57. Abularach SM, Xiong M, Kim S-Y, Bikovtseva AA, Butty VL, Schiavoni RP, et al. Human cartilage model of the precocious osteoarthritis-inducing COL2A1 p.Arg719Cys reveals pathology-driving matrix defects and a failure of the ER proteostasis network to recognize the defective procollagen-II. bioRxiv [Preprint]. 2024 Nov 7.
58. Yamada D, Takao T, Nakamura M, Lu M, Toguchida J, Takarada T. Establishment of high-throughput drug screening system using human limb bud mesenchymal cells derived from skeletal dysplasia-specific iPSC. In: Proceedings of the 95th Annual Meeting of the Japanese Pharmacological Society; 2022.
59. Nam Y, Rim YA, Jung SM, Ju JH. Cord blood cell-derived iPSCs as a new candidate for chondrogenic differentiation and cartilage regeneration. Stem Cell Res Ther. 2017 Jan 28;8(1):16.
60. Liu H, Yang L, Yu FF, Wang S, Wu C, Qu C, Lammi MJ, Guo X. The potential of induced pluripotent stem cells as a tool to study skeletal dysplasias and cartilage-related pathologic conditions. Osteoarthritis Cartilage. 2017 May;25(5):616-24.
61. Csobonyeiova M, Polak S, Nicodemou A, Zamborsky R, Danisovic L. iPSCs in Modeling and Therapy of Osteoarthritis. Biomedicines. 2021 Feb 12;9(2):186.
62. Khan NM, Diaz-Hernandez ME, Drissi H. Differentiation of Human Induced Pluripotent Stem Cells (iPSCs)-derived Mesenchymal Progenitors into Chondrocytes. Bio Protoc. 2023 Nov 5;13(21):e4874.
63. Nejadnik H, Diecke S, Lenkov OD, Chapelin F, Donig J, Tong X, et al. Improved approach for chondrogenic differentiation of human induced pluripotent stem cells. Stem Cell Rev Rep. 2015 Apr;11(2):242–53.
64. Lach MS, Wroblewska J, Kulcenty K, Richter M, Trzeciak T, Suchorska WM. Chondrogenic Differentiation of Pluripotent Stem Cells under Controllable Serum-Free Conditions. Int J Mol Sci. 2019 Jun 2;20(11):2711.
65. Ekholm J, Vukusic K, Brantsing C, Shaw G, Ur Rehman Bhatti F, Simonsson S, et al. Differentiation of Human Induced Pluripotent Stem Cells Toward Implantable Chondroprogenitor Cells. Cartilage. 2025 Jul 3:19476035251351713.
66. Eremeev A, Pikina A, Ruchko Y, Bogomazova A. Clinical Potential of Cellular Material Sources in the Generation of iPSC-Based Products for the Regeneration of Articular Cartilage. Int J Mol Sci. 2023 Sep 22;24(19):14408.
67. Khan NM, Diaz-Hernandez ME, Chihab S, Priyadarshani P, Bhattaram P, Mortensen LJ, et al. Differential chondrogenic differentiation between iPSC derived from healthy and OA cartilage is associated with changes in epigenetic regulation and metabolic transcriptomic signatures. Elife. 2023 Jan 30;12:e83138.
68. Lilianty J, Nur Patria Y, Stanley EG, Elefanty AG, Bateman JF, Lamandé SR. Generation of a heterozygous COL2A1 (p.R989C) spondyloepiphyseal dysplasia congenita mutation iPSC line, MCRIi001-B, using CRISPR/Cas9 gene editing. Stem Cell Res. 2020 May;45:101843.
69. Yamashita A, Morioka M, Kishi H, Kimura T, Yahara Y, Okada M, et al. Statin treatment rescues FGFR3 skeletal dysplasia phenotypes. Nature. 2014 Sep 25;513(7519):507–11.
70. Lee J, Taylor SE, Smeriglio P, Lai J, Maloney WJ, Yang F, et al. Early induction of a prechondrogenic population allows efficient generation of stable chondrocytes from human induced pluripotent stem cells. FASEB J. 2015 Aug;29(8):3399–410.
71. Takao T, Sato M, Fujisawa Y, Toyoda E, Yamada D, Hitsumoto Y, et al. A novel chondrocyte sheet fabrication using human-induced pluripotent stem cell-derived expandable limb-bud mesenchymal cells. Stem Cell Res Ther. 2023 Feb 24;14(1):34
72. Xiong J, Ma R, Xie K, Shan C, Chen H, Wang Y, et al. Recapitulation of endochondral ossification by hPSC-derived SOX9+ sclerotomal progenitors. Nat Commun. 2025 Mar 21;16(1):2781.
73. Hsueh YH, Buddhakosai W, Le PN, Tu YY, Huang HC, Lu HE, et al. Therapeutic effect of induced pluripotent stem cell -derived extracellular vesicles in an in vitro and in vivo osteoarthritis model. J Orthop Translat. 2022 Nov 8;38:141–55.
74. Bassett AR. Editing the genome of hiPSC with CRISPR/Cas9: disease models. Mamm Genome. 2017 Aug;28(7-8):348–64.
75. McTague A, Rossignoli G, Ferrini A, Barral S, Kurian MA. Genome Editing in iPSC-Based Neural Systems: From Disease Models to Future Therapeutic Strategies. Front Genome Ed. 2021 Mar 15;3:630600.
76. Dicks A, Wu CL, Steward N, Adkar SS, Gersbach CA, Guilak F. Prospective isolation of chondroprogenitors from human iPSCs based on cell surface markers identified using a CRISPR-Cas9-generated reporter. Stem Cell Res Ther. 2020 Feb 18;11(1):66.
77. De Kinderen P, Meester J, Loeys B, Peeters S, Gouze E, Woods S, et al . Differentiation of Induced Pluripotent Stem Cells Into Chondrocytes: Methods and Applications for Disease Modeling and Drug Discovery. J Bone Miner Res. 2022 Mar;37(3):397–410.
78. Ajalik RE, Alenchery RG, Cognetti JS, Zhang VZ, McGrath JL, Miller BL, et al. Human Organ-on-a-Chip Microphysiological Systems to Model Musculoskeletal Pathologies and Accelerate Therapeutic Discovery. Front Bioeng Biotechnol. 2022 Mar 14;10:846230.
79. Hwang JJ, Choi J, Rim YA, Nam Y, Ju JH. Application of Induced Pluripotent Stem Cells for Disease Modeling and 3D Model Construction: Focus on Osteoarthritis. Cells. 2021 Nov 5;10(11):3032.
80. Rodríguez Ruiz A, Dicks A, Tuerlings M, Schepers K, van Pel M, Nelissen RGHH, Freund C, Mummery CL, Orlova V, Guilak F, Meulenbelt I, Ramos YFM. Cartilage from human-induced pluripotent stem cells: comparison with neo-cartilage from chondrocytes and bone marrow mesenchymal stromal cells. Cell Tissue Res. 2021 Nov;386(2):309–20.
81. Lach MS, Rosochowicz MA, Richter M, Jagiełło I, Suchorska WM, Trzeciak T. The Induced Pluripotent Stem Cells in Articular Cartilage Regeneration and Disease Modelling: Are We Ready for Their Clinical Use? Cells. 2022 Feb 3;11(3):529.
82. Ali EAM, Smaida R, Meyer M, Ou W, Li Z, Han Z, et al. iPSCs chondrogenic differentiation for personalized regenerative medicine: a literature review. Stem Cell Res Ther. 2024 Jun 26;15(1):185.