Keywords
Haploinsufficiency, Mitochondrial diseases, MAVS, mRNA vaccine, POLG-related disorders, RotaTeq, Rotavirus, VP3
Background
Rotavirus (RV) is an agent of gastroenteritis and, to a lesser extent, neurological manifestations including seizures and epilepsy [1,2]. Before the development of effective vaccines, rotavirus was among the leading global drivers of life-threatening diarrhea in neonates and children less than five years of age [3]. Infectious rotavirus particles are triple-layered, non-enveloped virions housing a segmented, dsRNA genome consisting of eleven genomic segments that collectively encode 11-12 open-reading frames (ORFs), depending on the rotavirus strain [4,5]. Patients who fully recover from rotavirus infections exhibit neutralizing antibodies in convalescent plasma that are reactive to rotavirus VP4 and VP7, both of which comprise the rotaviral adhesion system on the outermost capsid layer of the infectious, triple-layered particle (TLP) [3,6]. Immunologists have exploited the segmented RV genome alongside RV host restriction to generate a wide berth of reassortant strains of rotavirus. In fact, RotaTeq is a pentavalent, live, attenuated rotavirus vaccine that contains five different rotavirus reassortant strains known as G1, G2, G3, G4, and P1 [7-9]. RotaTeq is especially effective when deployed, ranging from a 90-97% reduction in rotavirus-associated poor, clinical outcomes in the United States, the European Union, and Latin America [7,9]. However, the greatest incidence of pediatric rotavirus morbidity and mortality remains in the developing world, primarily in Africa, where delivery of RotaTeq has not been as successful [7,10].
Emerging virological evidence has revealed that non-human RV strains are capable of attenuating host mitochondrial antiviral signaling protein (MAVS) stability via rotavirus VP3 activity [11,12]. MAVS is well-characterized for its innate role in activating interferon expression as a host response upon the advent of the detection of viral ribonucleic acid signatures in the infected host cytosol [13-16]. However, MAVS is a multifunctional protein, and beyond its role in mediating innate cellular responses to viral infection, host MAVS is essential for maintaining normal mitochondrial functions [17]. For example, MAVS knockout mice exhibit a pleiotropic suite of abnormal mitochondrial deficiencies across numerous cell types, including disrupted cristae membranes, reduced electron transport chain (ETC) outputs, and attenuated oxidative phosphorylation rates [18-20]. Thus, during the course of rotavirus pathogenesis, some strains of rotavirus possess a VP3 configuration capable of driving MAVS degradation that blocks not only interferon expression but also normal mitochondrial activities [11]. Such mitochondrial toxicity by rotavirus VP3 needs to be carefully considered in patients afflicted with varying forms of mitochondrial diseases [21,22].
The POLG-related disorders are a hepatocerebral form of mitochondrial diseases [21,22]. The POLG locus encodes the catalytic subunit of the mitochondrial DNA (mtDNA) polymerase, and patients afflicted with POLG lesions typically present across a wide range of clinical manifestations such as cerebellar ataxia, epilepsy, seizures, neuropathy, myotonia, and acute liver failure or hepatopathy [21,23,24]. For patients diagnosed with mitochondrial diseases, excessive exposure to agents of mitochondrial toxicity is potentially life-threatening [25,26]. The recent findings that 1) non-human rotavirus VP3 can degrade human MAVS and 2) the absence of MAVS results in disrupted mitochondrial phenotypes collectively warrant not only a careful consideration of rotavirus infections in mitochondrial disease patients but may also suggest that the administration of the RotaTeq vaccine, which contains living rotavirus reassortant strains, could potentially be contraindicated for patients with POLG-related disorders.
Host defenses against RNA viruses utilize RLRs to drive interferon signaling
All human cells possess innate defenses against viral attack [27,28]. For instance, pattern recognition receptors (PRRs) such as the RIG-I-like receptors (RLRs) patrol the cytosolic compartment for evolutionarily-conserved pathogen-associated molecular pattern molecules (PAMPs) [29]. One such PAMP signature is the 5'-triphosphate moiety found on some viral RNAs that reveal themselves during virion uncoating following viral adhesion to and penetration of the host plasma membrane (Figure 1). Typically, viral proteases in the infectious virion proteolytically digest capsomere subunits from within the capsid to exert viral genome entry into the host cytosol. All viruses lack translation machinery and instead repurpose host ribosomes to manufacture viral proteins during the biosynthesis stages of the viral lifecycle. Viral biosynthesis stages involving viral protein synthesis generate not only viral peptide PAMPs but invariably produce viral mRNA PAMP signatures as well. In humans, RLRs such as RIG-I and MDA5 detect such viral RNA-based PAMPs [30,31]. Binding of viral RNA PAMPs to host RLR proteins induces a shape change revealing the protein domain known as CARD [29]. Subsequently, RLR CARDs form protein-protein interactions with other proteins harboring CARDs, including host MAVS (Figure 1). In the absence of viral infection, MAVS monomers are distributed along the periphery of the mitochondrial outer membrane (MOM) [29,32]. However, interaction with activated RLRs via CARDs permits post-translational modifications (PTMs) of MAVS via host E3 ubiquitin ligases, including TRIM31 (Figure 1). TRIM31 facilitates MAVS PTM by adducting K63-linked polyubiquitination tags which drives such modified MAVS monomers to oligomerize [14,33]. This multimeric complex of activated MAVS proteins serves as a scaffold for the recruitment and activation of cytosolic kinases, TBK1 and IKKε, both of which phosphorylate IRF3 and IRF7 transcription factors resulting in homodimerization or heterodimerization forms of the active transcription factor complexes (i.e., pIRF3/3, pIRF3/7, or pIRF7/7) [13,14,33,34]. Additionally, such a MAVS scaffold provides the opportunity to phosphorylate IKKβ at a phosphodegron site to initiate subsequent polyubiquitination for proteasome-mediated degradation [13,14,33]. Loss of cytosolic IKKβ levels relieves sequestration of NF-κB, and the latter is thus free to enter the nucleus alongside activated IRF3 and IRF7 to enhance the expression of interferons (IFNs) [13,14,33]. IFN secretion from such a virally-infected cell is perceived as a ligand by neighboring IFN receptor proteins found on nearby cells [27,35]. The paracrine consequences of successful IFN signal transduction in cells abutting the virally-infected cell includes the deployment of numerous antiviral responses (AVRs), placing a temporary moratorium on cellular DNA replication, RNA transcription, and protein translation activities [27,35]. When properly deployed, the interferon signal transduction system prevents cells located near the virally-infected cell from becoming ideal cellular hosts, and thus IFN signaling pathways interfere with viral lifecycles (Figure 1).
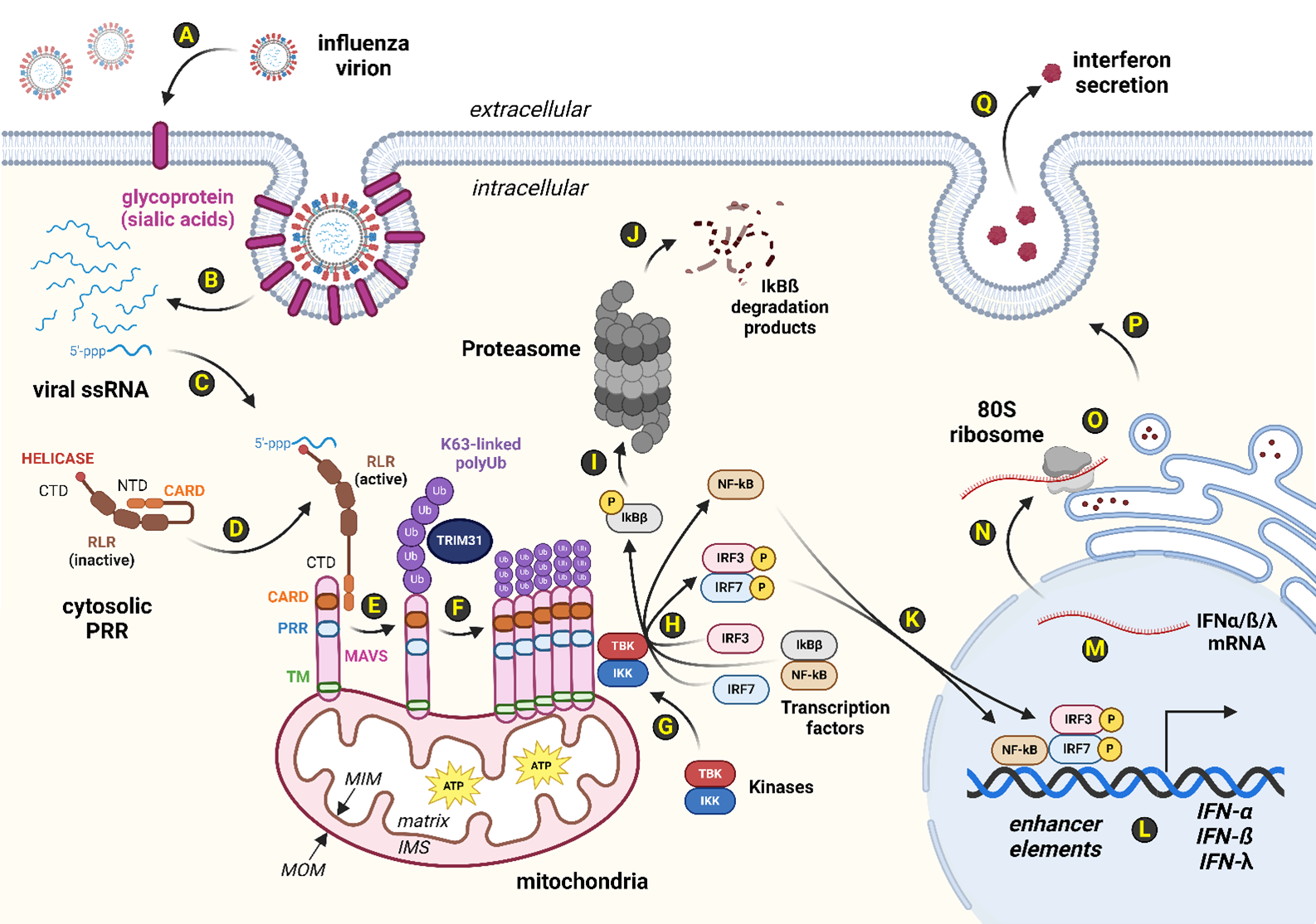
Figure 1. Host RLR-MAVS-IFN axis mediates innate defenses against RNA viral infection. Human cells innately leverage the interferon system to slow down viral propagation. Of the five types of host pattern recognition receptors (PRRs), the RLRs serve as cytosolic PRRs that detect viral RNA signatures. (A): Influenza virions use viral hemagglutinin (HA) as a spike protein to adhere to host glycoproteins that are decorated with sialic acid residues. (B): Uncoating of the viral capsid reveals the eight single-stranded, antisense RNAs (-ssRNAs) that comprise the segmented genome of influenza. (C): Many viral RNAs, including the -ssRNAs of influenza virus, contain a 5'-triphosphate moiety that triggers human RLR detection systems such as RIG-I and MDA5. (D): Human RLRs contain protein-protein binding domains known as CARDs on their amino-terminal domain (NTD) and a helicase domain on their carboxyl termini (CTDs) for binding viral RNAs found in the host cytosol. In the absence of viral RNAs, human RLRs exhibit a closed state where the RLR NTD interacts with the RLR CTD. (E): Upon binding of viral RNAs to RLRs, a shape change reveals their CARD domains which are free to complex with CARD domains found on human MAVS proteins. MAVS localizes to the mitochondrial outer membrane (MOM) via transmembrane (TM) domains found on the MAVS NTD. (F): RLR-bound MAVS are then post-translationally modified by TRIM31, an E3 ubiquitin ligase. Specifically, MAVS are modified via K63-linked polyubiquitin tags, typically at MAVS K10, K311, or K461. K63-linked polyubiquitinated MAVS monomers exhibit a conformation conducive to MAVS complex formation. (G): Oligomerized MAVS subunits serve as a scaffold for the recruitment of cytosolic kinases, including TBK1 and IKKε. (H): Cytosolic transcription factors, such as IRF3 and IRF7, are phosphorylated by TBK1 and IKKε kinases. Active, homodimeric or heterodimeric transcription factor complexes comprising pIRF3 and pIRF7 are thus able to form. In addition, (I) phosphorylation of a phosphodegron site on IKKβ shunts IKKβ towards polyubiquitination and eventual (J) proteasome degradation. (K): Cytosolic levels of IKKβ decrease, releasing the chelation system that keeps NF-κB trapped in the cytosol. Activated transcription factors, including NF-κB, pIRF3, and pIRF7, enter the nucleus to regulate gene expression by (L) enhancing transcription of interferon (IFN) genes. (M): IFN pre-mRNAs are processed into mature messages and (N) exit the nucleus via nuclear pores. (O): Host ribosomes bound to the rough endoplasmic reticulum (RER) translate IFN peptides into the RER lumen, commencing (P) endomembrane-mediated secretion to the (Q) extracellular pocket. Secreted IFN ligands serve to deploy transient suppression of replication, transcription, and translation services in nearby, adjacent cells. Abbreviations: 5'-ppp: 5' triphosphate moiety; 80S ribosome: Human ribosome; ATP: Adenosine triphosphate; CARD: Caspase activation and recruitment domains; CTD: Carboxyl-terminal domain; HA: Hemagglutinin; IFN: Interferon; IFN-α: Type 1 IFN; IFN-ß: Type 1 IFN; IFN-λ: Type 3 IFN; IKKβ: Inhibitor of nuclear factor kappa-B kinase subunit beta; IKKε: Inhibitor of nuclear factor kappa-B kinase subunit epsilon; IMS: Intermembrane space; IRF3: Interferon response factor 3; IRF7: Interferon response factor 7; K63-linked polyUb: Post-translational modification that activates MAVS oligomerization; MAVS: Mitochondrial antiviral-signaling protein; MDA5: Melanoma differentiation-associated protein 5; MIM: Mitochondrial inner membrane; MOM: Mitochondrial outer membrane; mRNA: Messenger RNA; NF-κB: Nuclear factor kappa B; NTD: Amino terminal domain; pIRF3: Phosphorylated IRF3; pIRF7: Phosphorylated IRF7; PRR: Pattern recognition receptor as protein; PRR: Proline-rich region as domain; RIG-I: Retinoic acid-inducible gene I; RLR: RIG-I-like receptor; RNA: Ribonucleic acid; ssRNA: Single-stranded RNA; -ssRNA: Negative or antisense ssRNA; TBK1: TANK-binding kinase 1; TM: Transmembrane domain; TRIM31: Tripartite motif-containing protein 31; Ub: Ubiquitin. Image created via BioRender.
Rotavirus infection attenuates host defenses by driving MAVS degradation
Although non-human rotaviruses do not typically drive poor clinical outcomes in healthy human patients, these rotaviruses still execute an attenuated lifecycle during human infection [6,36]. RV TLPs use VP4 and VP7 peptides to adhere to host cells via binding to host sialoglycan-decorated glycoproteins or histo-blood group antigens (HBGAs) found on the target plasma membrane (Figure 2) [37]. Unlike most viruses, the rotavirus does not fully uncoat following viral penetration of a suitable host cell [3,5]. Instead, the outer layer of the RV TLP fuses with the endosomal membrane allowing the escape of a RV double-layered particle (DLP) from the endosomal lumen into the cytosolic pocket [3,5]. In this fashion, any PAMPs derived from the eleven dsRNA RV genomic segments are protected from direct contact by host RLR detection systems (Figure 2) [3,5]. Additionally, the RV VP1 found within the DLP has RNA-dependent RNA polymerase (RdRp) activity [3,5]. From the insulated vantage of a rotavirus DLP, VP1 extrudes nascent RV mRNA molecules from the DLP into the host cytosol to drive the rotaviral lifecycle (Figure 2] [3,5]. One such viral message is RV VP3, which encodes a multi-functional protein with at least four distinct active sites [12,38,39]. Recent evidence has demonstrated that RV VP3 can bind to human MAVS in virally-infected HT-29 cells (i.e., a human colorectal epithelial cell line] [11]. RV VP3 protein likely facilitates the K48-linked polyubiquitination of host MAVS monomers by host TRIM25, an E3 ubiquitin ligase (Figure 2) [11]. Such MAVS PTMs shunt MAVS to the host proteasome which effectively degrades cellular MAVS levels [11]. In fact, human MAVS levels in human HT-29 cells infected by either simian RV SA11, simian RV RRV, or porcine RV OSU strains exhibit a VP3-dependent MAVS protein level reduction by 70-80% [11]. The consequences of MAVS degradation by rotavirus VP3 are profound. From an immunological perspective, loss of MAVS results in failure to activate key transcription factors for interferon expression; the rotavirus-infected cell is thus unable to secrete interferon, and consequently unable to signal to adjacent cells to express interferon-stimulated genes (ISGs) for paving antiviral responses ahead of future virion infection events [40-42]. From a bioenergetics perspective, the degradation of MAVS disrupts the overall function of the afflicted mitochondrial organelle [18-20]. Numerous lines of evidence from mouse MAVS knockout data collectively point to the key role that host MAVS plays in normal mitochondrial activities, including the operation of electron transport chains to sustain adequate ATP output via oxidative phosphorylation [18-20]. Thus, as rotavirus infection destabilizes host MAVS, mitochondrial toxicity likely ensues, and cellular starvation pathways (e.g., AMPK signaling) activate to drive mitochondrial organelle renewal [43-45]. As each mitochondrion typically requires 2-10 copies of mtDNA, and a single eukaryotic cell might harbor up to 1,000 total mitochondria, the extent of mitochondrial toxicity by rotavirus pathogenesis can be potentially expansive [46]. Expression of the POLG1 and POLG2 loci, which encode the catalytic and scaffold subunits of the mtDNA polymerase complex, respectively, would thus be extensively drawn upon to satiate the cellular demands to reassert bioenergetic homeostasis during the lifecycle of rotavirus in an afflicted cell (Figure 2).
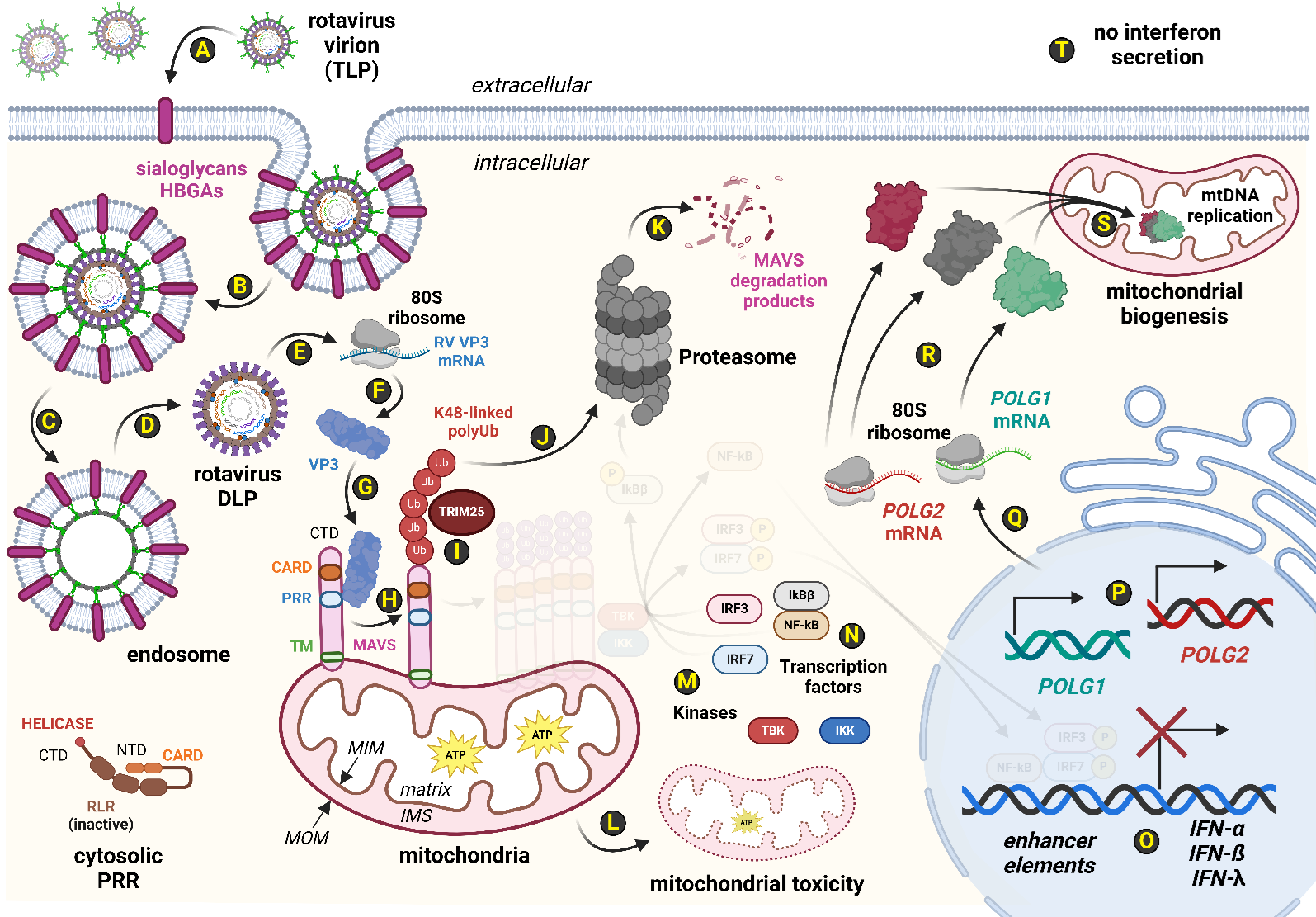
Figure 2. Rotavirus infection attenuates host defenses while driving mitochondrial toxicity. The rotavirus (RV) lifecycle employs several molecular means of bypassing innate host RLR systems. (A): The rotavirus virion is a triple-layered particle (TLP), leveraging viral VP4 and VP7 proteins to adhere to host sialoglycans and human blood group antigens (HBGA)s. (B-D): Endocytosis of the rotavirus TLP commences, and the outermost layer containing VP4 and VP7 is left behind in the endosomal membrane during egress of rotavirus double-layered particles (DLP) into the host cytosol. (E): Of note, the 11 segments of the rotaviral double-stranded RNA (dsRNA) genome are not exposed to the cytosol but rather are retained within the DLP. This reduces the ability of cytosolic RLR systems such as host RIG-I and MDA5 to detect RV infection. Viral mRNAs are synthesized within the DLP and these viral mRNAs, including that of the RV VP3 message, exit the DLP for the cytosol where they are (F) translated into viral proteins. (G): Some strains carry an RV VP3 that can bind to host MAVS at a SPLTSS motif near the proline-rich region (PRR) of MAVS. (H): VP3-bound MAVS are believed to be post-translationally modified by K48-linked polyubiquitin tags facilitated by the (I) E3 ubiquitin ligase known as TRIM25. Unlike K63-polyUb modifications, K48-polyubiquitination of host MAVS shunts modified MAVS to the (J-K) proteasome for MAVS degradation. (L): MAVS degradation by the proteasome disrupts mitochondrial integrity, oxidative phosphorylation output, and overall function. (M-O): Additionally, MAVS degradation by the proteasome prevents the MAVS oligomerization scaffold critically needed by cytosolic kinases, TBK1 and IKKβ, for complex formation. Thus, transcription factors (IRF3, IRF7 and NF-κB) remain cytosolic which greatly diminishes IFN gene expression. (P-R): Meanwhile, damage to mitochondrial productivity rates of ATP requires POLG1 and POLG2 expression to form the mitochondrial DNA (mtDNA) polymerase, a heterotrimeric complex comprising one catalytic subunit copy of POLG1 and two scaffold subunits of POLG2. (S): A single human cell may have up to a thousand mitochondria, and each mitochondrial organelle typically contains 2-10 copies of mtDNA chromosomes. The mtDNA polymerase complex assembles in the mitochondrial matrix where mtDNA synthesis commences during mitochondrial biogenesis. (T): While the cell reasserts homeostasis over mitochondrial toxicity resulting from RV infection, the infected cell is unable to communicate its virally-infected state to neighboring cells due to loss of IFN secretion. Abbreviations: 5'-ppp: 5' triphosphate moiety; 80S ribosome: Human ribosome; ATP: Adenosine triphosphate; CARD: Caspase activation and recruitment domains; CTD: Carboxyl-terminal domain; DLP: Double-layered particle; DNA: Deoxyribonucleic acid; dsRNA: Double-stranded RNA; HBGA: Histo-blood group antigen; IFN: Interferon; IFN-α: Type 1 IFN; IFN-ß: Type 1 IFN; IFN-λ: Type 3 IFN; IKKβ: Inhibitor of nuclear factor kappa-B kinase subunit beta; IKKε: Inhibitor of nuclear factor kappa-B kinase subunit epsilon; IMS: Intermembrane space; IRF3: Interferon response factor 3; IRF7: Interferon response factor 7; K48-linked polyUb: Post-translational modification that degrades MAVS monomers; MAVS: Mitochondrial antiviral-signaling protein; MDA5: Melanoma differentiation-associated protein 5; MIM: Mitochondrial inner membrane; MOM: Mitochondrial outer membrane; mRNA: Messenger RNA; mtDNA: Mitochondrial DNA; NF-κB: Nuclear factor kappa B; NTD: Amino terminal domain; POLG: Gene encoding the catalytic subunit of mtDNA polymerase; POLG1: Alias for POLG; POLG2: Gene encoding the scaffold subunits of mtDNA polymerase; PRR: Pattern recognition receptor as protein; PRR: Proline-rich region as domain; RIG-I: Retinoic acid-inducible gene I; RLR: RIG-I-like receptor; RNA: Ribonucleic acid; RV: Rotavirus; SPLTSS: MAVS motif for binding by RV VP3; TBK1: TANK-binding kinase 1; TLP: Triple-layered particle; TM: Transmembrane domain; TRIM25: Tripartite motif-containing protein 25; Ub: Ubiquitin; VP3: RV Viral protein 3; VP4: RV Viral protein 4; VP7: RV Viral protein 7. Image created via BioRender.
Patients afflicted with POLG-related disorders are susceptible to mitochondrial toxicity
Mitochondrial DNA depletion syndromes (MDDS) present across a wide spectrum of clinical manifestations [21,22,24]. POLG-related disorders are a form of MDDS in which neuronal and hepatic systems are frequently affected [21,23]. Although typically two null POLG lesions in trans are required for phenotypic presentation, there are examples of heterozygous carriers of single pathogenic variants that exhibit hepatocerebral MDDS manifestations [24,47]. Patients diagnosed with POLG-related disorders are especially sensitive to agents of mitochondrial toxicity, which can include certain medications such as aminoglycoside antibiotics that interfere with mitochondrial ribosomes in the mitochondrial matrix or antileptics like valproate sodium that can drive life-threatening, acute liver failure [48-50]. Additionally, many infectious viral agents of disease can damage host mitochondria during viral infections, including human respiratory syncytial virus (RSV), SARS-CoV-2, hepatitis viruses (i.e., HBV and HCV), and measles virus [51]. Most of these viral agents leverage a molecular attack against normal host MAVS functions, often driving MAVS degradation [51]. Taken together, mitochondrial toxicity from contraindicated medications or certain viral agents might exacerbate clinical manifestations in POLG patients as mtDNA polymerase activities become overwhelmed. As rotavirus VP3 from certain rotavirus strains can degrade MAVS, this raises the question as to whether the living rotavirus reassortant strains found in the pentavalent RotaTeq vaccine are safe for patients harboring POLG lesions.
Results
Orthologous VP3 peptide sequences exhibit high amino-terminal sequence conservation
Although the five living RV reassortant strains found in the RotaTeq vaccine cocktail have been extensively sequenced and characterized at the molecular level, the effect of each reassortant strain's VP3 locus on host MAVS levels during RotaTeq-mediated infection has not yet been investigated [8]. Simian and porcine strains of rotavirus carry VP3 that can reduce human MAVS protein levels by ~70% [11]. Additionally, molecular deletion analyses reveal that the VP3 amino-terminal domain (NTD), spanning the first 171 amino acid residues, is sufficient to drive host MAVS degradation in infected human cell lines [12]. Comparative analyses of VP3 NTD peptide sequences across RotaTeq reassortants as well as VP3 NTDs from non-human rotavirus strains (i.e., simian and porcine) known to degrade human MAVS reveals exceptionally high (>90%) peptide sequence conservation (Table 1). A complete, pairwise comparison of all VP3 NTDs from MAVS-degrading RV strains (i.e., CaRV SA11 and SsRV OSU), a known non-MAVS-degrading RV strain (i.e., HsRV Wa), and all five RotaTeq reassortant strains (i.e., G1, G2, G3, G4, and P1) is provided in Supplementary File 1.
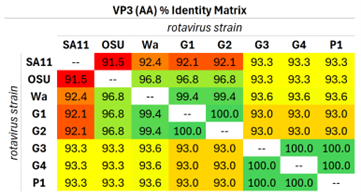
Table 1. Orthologous RV VP3 NTDs exhibit exceptional amino acid conservation. Eight VP3 NTDs representing VP3 residues 1-171 were aligned in pairwise fashion using JalView and JABAWS software. A total of 28 (8 termial or the 8th triangular number) unique pairwise comparisons were constructed for each amino acid residue position of the organismal VP3 NTDs. VP3 NTD peptide sequences for simian RV SA11 and porcine RV OSU strains exhibited high conservation (i.e., >90%) to all other organismal VP3 NTDs. The RotaTeq vaccine is a pentavalent vaccine comprising five reassortant RV strains derived from bovine RV and human RV strains. From the perspective of the VP3 locus, RotaTeq's VP3 peptide sequence shares a high degree of conservation of the VP3 NTD to those derived from strains empirically capable of degrading human MAVS during host rotaviral infection, including the VP3 NTDs of simian RV SA11 and porcine RV OSU [11]. Abbreviations: RV: Rotavirus; RV OSU: Porcine rotavirus strain; RV SA11: Simian rotavirus strain; RV Wa: Human rotavirus strain; G1, G2, G3, G4, P1: Rotavirus reassortant strains found in the RotaTeq vaccine.
Phylogenetic analyses reveal close VP3 peptide sequence relationships in many rotavirus strains
Cladistics for either 1) full-length VP3 (VP3-FL) or 2) the VP3 NTD alone collectively showcase that the VP3 loci found in the RotaTeq G1 and G2 reassortant strains share a more recent common ancestor to human rotavirus strain Wa (Figure 3). Notably, human rotavirus Wa VP3 is unable to degrade host MAVS in RV-infected HT-29 cells [11,12]. Dendrogram analyses thus suggest that the RotaTeq G1 VP3 and RotaTeq G2 VP3 loci are likely not contraindicated to POLG patients (Figure 3). However, the VP3 evolutionary relationship between bovine rotavirus (BtRV) strains (i.e., BtRV BrB-9, WI78-8, and WI79-4), comprising three reassortant strains of the RotaTeq vaccine (i.e., G3, G4 and P1), and either simian RV SA11, porcine RV OSU, and several human rotavirus strains is unclear and polyphyletic (Figure 3). As phylogeny fails to place BtRV VP3-FL or VP3-NTD peptide sequences near non-MAVS-degrading HsRV Wa strains, caution is therefore warranted when providing RotaTeq to mitochondrial disease patients, including those afflicted by POLG-related disorders (Figure 3). Although orthologous RV VP3 phylogeny at the peptide level (i.e., full-length and NTD sequences) yields polyphyletic dendrograms, analysis of RV VP1 evolution yields numerous monophyletic clades centered around the host range of rotavirus strains (Supplementary File 2). This is likely due to a higher stringency of evolutionary pressure to remain constant at the VP1 locus, which encodes the RNA-dependent RNA polymerase enzyme that is essential for the rotavirus lifecycle. Notably, all VP1 sequences found in the RotaTeq vaccine exhibit evolutionary proximity to the bovine RV WC3 strain on account of the parental strains used during reassortant construction. Taken together, VP1 exhibits a cladistics pattern indicative of host restriction likely due to stringent evolutionary pressure for constancy, while it is unclear as to the degree by which the BtRV VP3 peptides found within the RotaTeq formulation (i.e., G3, G4, and P1) relate with respect to known MAVS-degrading VP3 sequences (e.g., RV-SA11-VP3, and RV-OSU-VP3).
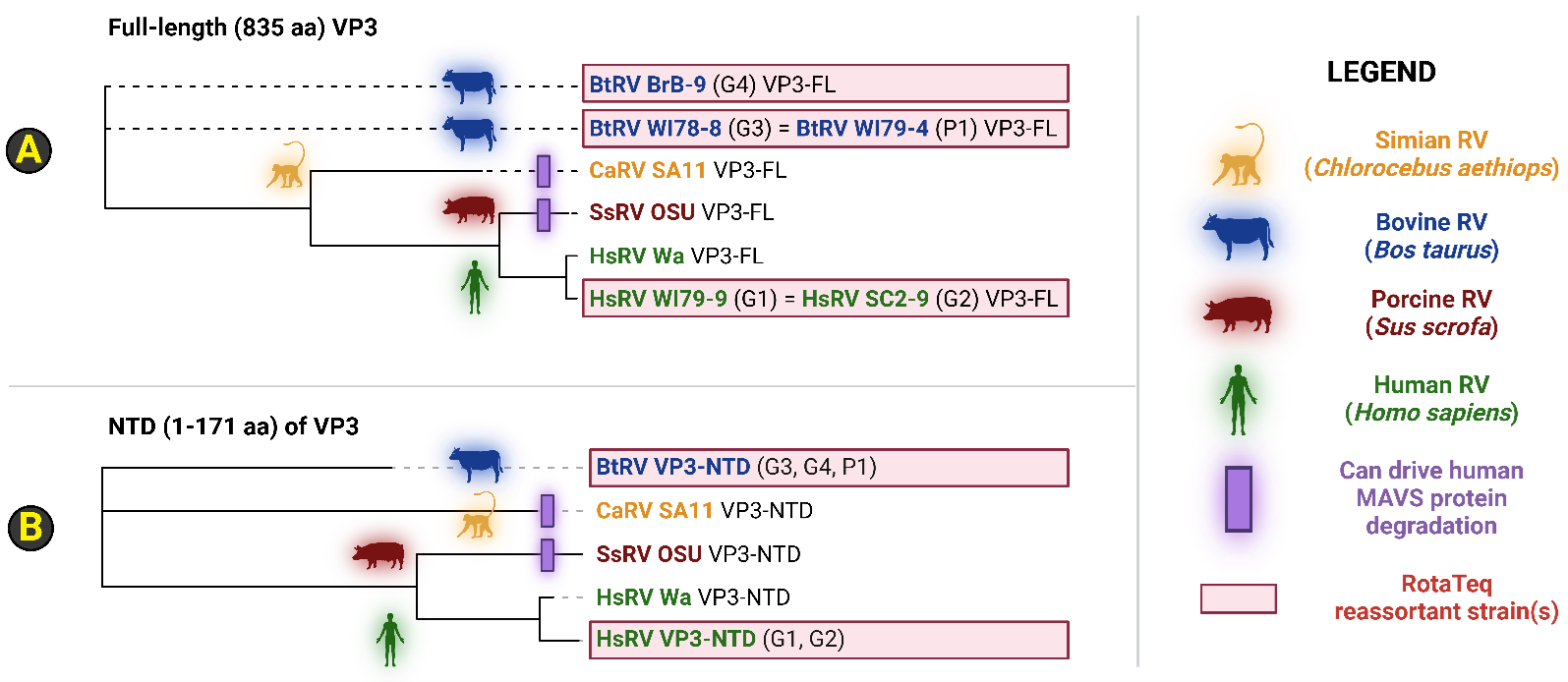
Figure 3. Phylogenetic analysis of the rotavirus reassortant VP3 peptide sequences found in the RotaTeq vaccine. Dendrograms were generated for either (A) full-length VP3 or (B) only VP3 NTDs from a total pool of 8 VP3 peptide sequences, spanning four host organisms. The RotaTeq vaccine contains five distinct reassortant strains that harbor a RV VP3 gene permutation derived from bovine RV and human RV strains. Simian RV and porcine RV strains possess an orthologous VP3 capable of bringing about the degradation of human MAVS during RV infection of human colorectal epithelial cell lines. Of five VP3 genes found in the pentavalent RotaTeq reassortant vaccine, two VP3 sequences derive from human rotavirus strains, while the remaining three trace parental origins to bovine rotavirus strains. The VP3 of human RV Wa strain is unable to drive MAVS degradation during human RV infection of human cells, and both human RV VP3 peptides found in the RotaTeq vaccine exhibit the greatest similarity to human RV Wa sequences. However, it is unclear if the three VP3 peptide sequences derived from reassortants involving a bovine origin are capable of mediating MAVS degradation. Abbreviations: AA: Amino acid(s); BtRV: Bos taurus RV; CaRV: Chlorocebus aethiops RV; HsRV: Homo sapiens RV; MAVS: Mitochondrial antiviral-signaling protein; NTD: Amino-terminal domain; RV: Rotavirus; SsRV: Sus scrofa RV; VP3: RV Viral protein 3; VP3-FL: Full-length VP3 peptide; VP3-NTD: VP3 peptide segment from 1-171 aa. Image created via BioRender.
Discussion
The RotaTeq vaccine may be contraindicated for mitochondrial disease patients
Globally, approximately 600,000 deaths in children under five years of age are attributed to gastroenteritis caused by rotavirus, with disease burden carried mostly by developing countries [3,52,53]. Administration of the RotaTeq vaccine is a significant boon in the prophylactic arsenal against rotavirus pathogenesis, reducing not only pediatric mortality rates but also many measurable facets of rotavirus disease burden [7-9]. Examples include reductions in rotavirus-associated emergency-department visits and hospitalizations, as well as greatly diminishing rotavirus gastroenteritis disease and severity [7-9]. However, in any given population, there may exist a rare cohort that cannot receive vaccine doses for a variety of reasons. In the case of persons carrying a single, pathogenic POLG variant or patients diagnosed with POLG-related disorders, mitochondrial toxicity concerns might exist during exposure to living rotavirus strains that possess a VP3 capable of degrading host MAVS [21,24,47]. Thus, further research into the effects of MAVS degradation by the RV VP3 possessed by each of the five reassortant strains (i.e., G1, G2, G3, G4 and P1) comprising the RotaTeq vaccine would be warranted on the behalf of mitochondrial disease patients. Mitochondrial disease patients, including those afflicted with POLG-related disorders, are especially vulnerable to mitochondrial toxicity. Perhaps the most dramatic clinical contraindication case involves acute liver failure and sudden lethality in POLG patients who received the antileptic known as valproate sodium [48–50]. In human cells, valproate treatment induced elevated rates of mitochondrial biogenesis which were not sustainable for human POLG mutant cell lines [54]. While valproate sodium represents a severe contraindication for POLG patients, the degree to which other agents of mitochondrial toxicity drive poor clinical outcomes is not systematically and fully characterized. Clinical guidelines do exist however to restrict known medications that are known to exhibit varying degrees of mitochondrial toxicity, including the aminoglycoside class of antibiotics (e.g., streptomycin, erythromycin, etc.) and various antileptics [48-50].
If RotaTeq's reassortant strains can degrade MAVS, we recommend three different strategies to minimize contraindications and complications in patients afflicted with POLG-related disorders (Figure 4) [26,55,56]. The simplest strategy would be to incorporate POLG genotype surveillance prior to RotaTeq administration [57,58]. RotaTeq vaccine might then be withheld from mitochondrial disease patients who exhibit POLG heterozygosity (i.e., carriers) or those who are POLG-/- (i.e., homozygotes, or compound trans heterozygotes) until empirical evidence exists that the five RotaTeq reassortant VP3 peptides are incapable of mediating the degradation of host MAVS protein levels in human cells during RotaTeq (i.e., rotavirus) infection (Figure 4A-B). In such a scenario, herd immunity granted by POLG+/+ individuals who received the RotaTeq vaccine would offer passive protection to mitochondrial disease cohorts (Figure 4B). If future work empirically demonstrates that RotaTeq exhibits a reassortant VP3 capable of eliciting host MAVS degradation during the rotavirus lifecycle in RotaTeq-infected human cells, then a recombinant RotaTeq vaccine can be designed that leverages replacement of all five VP3 genetic loci across the five RotaTeq reassortant strains with the non-MAVS degrading VP3 possessed by the HsRV Wa strain (Figure 4C) [11]. The HsRV Wa VP3 lacks the ability to degrade human MAVS in human colorectal epithelial cells (i.e., HT-29), and this locus would be an ideal gene candidate for a recombinant RotaTeq vaccine design [11]. Such a recombinant RotaTeq vaccine could be administered to any patient without prior knowledge of their POLG locus, as this vaccine design should not exhibit mitochondrial toxicity. Lastly, an mRNA delivery package might be better suited for replacing the reassortant-centric vaccine design that embodies the RotaTeq strategy. An mRNA vaccine against human rotavirus type A should minimally contain VP4 and VP7 immunogenicity. Extending upon the RotaTeq approach, five different VP4 peptides and five different VP7 peptides can be delivered in a decavalent mRNA vaccine cocktail comprising all the VP4 and VP7 ORFs embodied by the RotaTeq vaccine (Figure 4D; Supplementary File 3). This proposed rotavirus mRNA vaccine design should mirror the efficacy of RotaTeq's reassortant approach, namely via the attenuated RV TLP delivery of exogenous VP4 and VP7 antigen to stimulate host production of neutralizing antibodies, with the added advantage of a simplified manufacturing process agnostic of viral reassortant strain production.
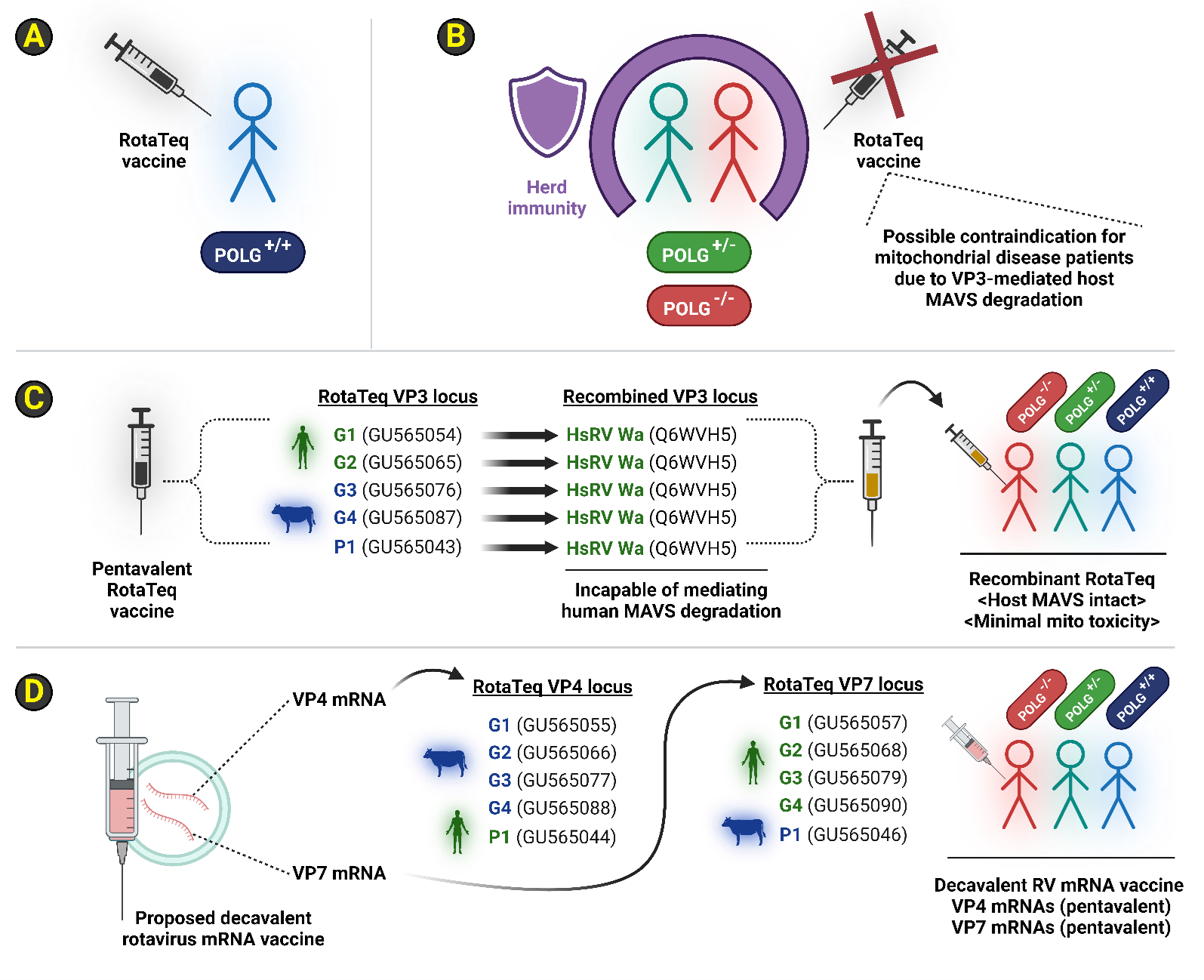
Figure 4. Proposed vaccination approaches for mitochondrial disease patients, including those afflicted with POLG-related disorders. Three overall strategies are proposed here for maximizing rotaviral vaccine efficacy in the context of mitochondrial disease patients exhibiting lesions at POLG1 (aka POLG). (A-B): RotaTeq is only given to patients that do not exhibit any pathogenic variants at the POLG locus until biochemical and viral infection assays can rule out bovine RV VP3 from degrading human MAVS during infection of human cells. By restricting the RotaTeq vaccine to only wild-type (WT) POLG+/+ individuals, herd immunity is extended to POLG+/- carriers (heterozygotes) or POLG-/- patients due to mitochondrial toxicity contraindication concerns. Note that haploinsufficiency manifestations do exist in the literature for POLG heterozygotes likely due to either POLG genotoxicity or agents of mitochondrial toxicity. (C): Alternatively, the RotaTeq vaccine might be modified via recombinant DNA technology with the overall goal of replacing all five VP3 genes currently found in the RotaTeq vaccine with the VP3 gene harbored by the human RV Wa strain. The human RV Wa VP3 empirically demonstrates an inability to facilitate host MAVS degradation during RV infection of human cell culture. Such a recombinant, pentavalent RotaTeq vaccine formulation could thus be safely given to all patients without requiring POLG locus screening. This minor modification still retains all the VP4 and VP7 immunogenicity found in the original RotaTeq formulation and should thus track the exemplary vaccine effectiveness of RotaTeq vaccine protection against poor clinical outcomes by rotavirus infections. RV VP3 sequence accessions for GenBank or UniProt are shown in parentheses. (D): Generating an mRNA vaccine cocktail derived solely from VP4 and VP7 immunogenicity removes VP3 entirely from the equation. Each of the precise VP4 and VP7 peptide sequences and thus open-reading frames (ORFs) are already known across all five bovine/human RV reassortants. A cocktail containing five VP4 ORFs and five VP7 ORFs delivered as a heterogenous mixture of 10 total mRNA molecules should not be contraindicated to mitochondrial disease patients and should thus minimize risk when administered to any patient without prior knowledge of their POLG genotype. The production advantage of an mRNA vaccine approach might additionally simplify the significant virological methodology required to sustain the precise reassortant structures as currently defined in the RotaTeq formulation. RV VP4 and RV VP7 sequence accessions for GenBank are shown in parentheses. Abbreviations: MAVS: Mitochondrial antiviral-signaling protein; mRNA: Messenger RNA; ORF: Open reading frame; POLG: Gene encoding the catalytic subunit of mtDNA polymerase; POLG-/-: Double loss-of-function genotype for the POLG locus; POLG+/-: Heterozygous POLG genotype; POLG+/+: WT genotype for the POLG locus; POLG1: alias name for POLG; RNA: Ribonucleic acid; RV: Rotavirus; VP3: RV Viral protein 3; WT: Wild-type or normal. Image created via BioRender.
Lastly, we wish to remind the reader that all methods in this paper are purely computational and bioinformatics by nature and thus lack empirical validation. Our bioinformatics analyses are ultimately unable to classify RotaTeq VP3 NTD as similar to 1) known MAVS-degrading CaRV SA11 VP3, 2) known MAVS-degrading SsRV OSU VP3, or 3) known non-MAVS-degrading HsRV Wa VP3 (Figure 3). Due to this uncertainty, we urge caution with respect to RotaTeq administration in POLG patients (Figure 4A) and suggest a vaccine reformulation strategy (Figure 4B-4C) only if future work empirically demonstrates that RotaTeq VP3 peptides are capable of degrading human MAVS in POLG patients.
Methods
A pipeline overview of all computational biology, data science, and bioinformatics methodologies employed in this publication is illustrated in Figure 5. Broadly speaking, this workflow involves procurement for DNA and protein sequences, alignment operations, sequence identity matrices, dendrogram generation, and mRNA vaccine design (Figure 5).
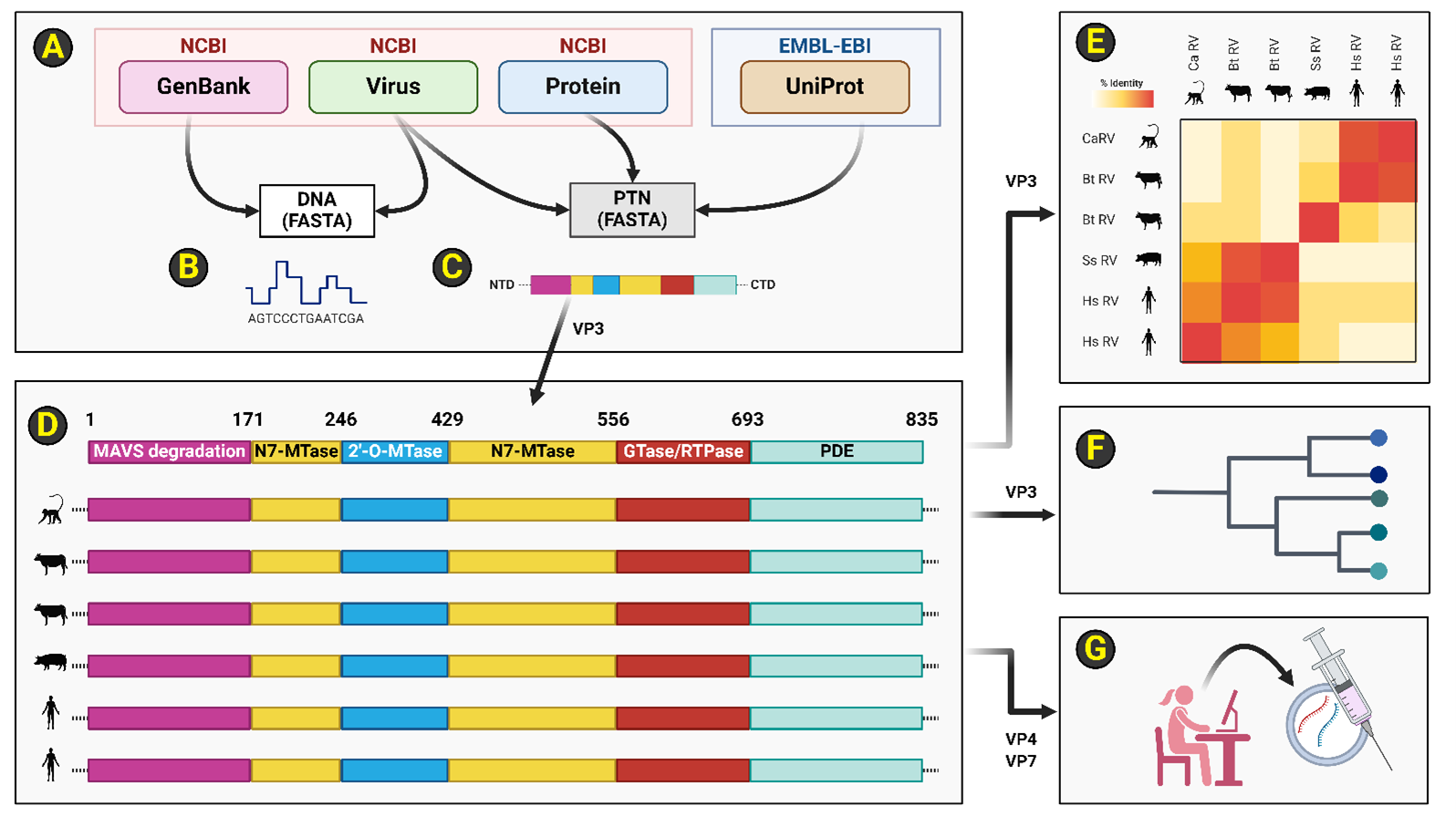
Figure 5. Schema of employed bioinformatics modules and overall analytical workflow. (A): DNA and peptide sequences were obtained from public-domain databases administered by NCBI or EMBL-EBI. NCBI GenBank entries were accessed for all RotaTeq reassortant strains (i.e., G1, G2, G3, G4 and P1) for VP1, VP3, VP4, and VP7 loci. Accessions from NCBI Virus and the EMBL-EBI UniProt databases were utilized for simian and porcine RV strains for the VP1 and VP3 loci. The NCBI Protein database was accessed for bovine RV VP1 sequences. Custom BioPython scripts leveraging the Seq class were written to organize and wrangle DNA and protein sequences for each organismal accession. (B-C): DNA and peptide sequences were obtained in FASTA file format from indicated databases. (D): An example subset of the primary peptide sequences of organismal RV strain VP3 is illustrated here. The orthologous RV VP3 peptide sequences comprise five protein domains (i.e., MAVS degradation, N7-MTase, 2'-O-MTase, GTase/RTPase, and PDE) across a primary peptide sequence of 835 residues. RV VP3 domains are demarcated by residue number as well as color bars. Organismal RV VP3 peptide sequences were loaded as full-length or VP3-NTDs (i.e., residues 1-171) into JalView software and alignments were performed using JABAWS and ClustalOmega. (E): Custom BioPython scripts were written to perform every unique pairwise comparison at a per-residue resolution for VP3-NTD across all organismal RV VP3 accessions in FASTA format. Heatmaps representing percent amino acid identity were then calculated for a total of 8 termial comparisons which equates to 28 total unique pairwise RV VP3 NTD comparisons. (F): Dendrograms were generated for RV VP1, RV VP3-FL, and RV VP3-NTD peptide sequences. For each dendrogram, Newick trees in NHX format were created using the default OneClick workflow in the NGphylogeny.fr pipeline which sequentially leverages bioinformatics packages BMGE, FastME, MAFFT, and Newick utilities in a total of fifteen overall steps on a Galaxy server. Graphical visualization of each dendrogram (i.e., VP1, VP3, or VP3-NTD) was performed using iTOL and categorical annotations were added using BioRender. (G): A non-VP3 based mRNA rotavirus vaccine to limit potential contraindications in POLG patients was computationally designed. NCBI GenBank accessions regarding VP4 and VP7 sequences from all five RotaTeq reassortant strains were sourced as immunogen material for a decavalent mRNA rotavirus vaccine package. The EMBL-EBI EMBOSS Backtranseq method was employed to reverse translate VP4 and VP7 peptide sequences into an open reading frame replete with human codon optimization parameters. Each of ten ORFs were placed into a vehicle based on the published Moderna SARS-CoV-2 mRNA vaccine sequences. All elements, including the signal sequence were retained to ensure host cell production of exogenous antigens (i.e., via host endomembrane secretion systems) to drive a humoral vaccine strategy that favors a neutralizing antibody response against the RV virion's adhesion system (i.e., VP4 and VP7 of the RV TLP). Abbreviations: 2'-O-Mtase: Ribose-2'-O-methyltransferase; BMGE: Block Mapping and Gathering with Entropy; CTD: Carboxyl-terminal domain; DNA: Deoxyribonucleic acid; EBI: European Bioinformatics Institute; EMBL: European Molecular Biology Lab; EMBOSS: European Molecular Biology Open Software Suite; FASTA: A file format for handling sequence information; FastME: Fast Minimal Evolution; G1: RotaTeq reassortant strain; G2: RotaTeq reassortant strain; G3: RotaTeq reassortant strain; G4: RotaTeq reassortant strain; GTase: Guanylyl-transferase; iTOL: Interactive Tree of Life; JABAWS: Java Bioinformatics Analyses Web Service; MAFFT: Multiple Alignment using Fast Fourier Transform; N7-Mtase: Guanine-N7-methyltransferase; NCBI: National Center for Biotechnology Information; NHX: A file format for cladistics; NTD: Amino-terminal domain; ORF: Open reading frame; P1: RotaTeq reassortant strain; PDE: Phosphodiesterase; POLG: Encodes the catalytic subunit of the mtDNA polymerase complex; RV: Rotavirus. Image created via BioRender.
Data management
VP1, VP3, VP4, and VP7 nucleic acid and peptide sequences were obtained from either NCBI's GenBank or EMBL-EBI's UniProt databases [59-63]. For RotaTeq reassortant strains (i.e., G1, G2, G3, G4 and P1), the following GenBank sequences were analyzed: G1-VP1 (GU565052), G2-VP1 (GU565063), G3-VP1 (GU565074), G4-VP1 (GU565085), P1-VP1 (GU565041), G1-VP3 (GU565054), G2-VP3 (GU565065), G3-VP3 (GU565076), G4-VP3 (GU565087), P1-VP3 (GU565043), G1-VP4 (GU565055), G2-VP4 (GU565066), G3-VP4 (GU565077), G4-VP4 (GU565088), P1-VP4 (GU565044), G1-VP7 (GU565057), G2-VP7 (GU565068), G3-VP7 (GU565079), G4-VP7 (GU565090), and P1-VP7 (GU565046) [8]. For the simian RV SA11 strain, the following peptide sequences were examined: SA11-VP1 (UniProt: A2T3S0, O37061, and P22678; NCBI Virus: ABE66463 and CAA34732), and SA11-VP3 (UniProt: A2T3S5). For porcine RV strains, the following peptide sequences were analyzed: VP1 (NCBI Virus: WRK24342, WRK24353, WIG65342, UZX50353, QZA75558, QZA75559, AYA93300, AYA93301, AYA93327, AYA93328, QCE32082, QCC26610, AYP00126, AXS77342, AVR65453, AQQ12223, AQQ12224, AQQ12225, AQQ12226, AQQ12227, ANQ45168, BAQ95493, BAQ95504, BAQ95515, BAP28211, AGP05014, AEO79879, AEL20332, AEL20333, AEL20334, AEL20335, and ADF57909), and OSU-VP3 (UniProt: Q6WNV8). For the human RV WA strain, the following UniProt peptide sequences were utilized: WA-VP1 (A4ZCW8), and WA-VP3 (Q6WVH5). The bovine RV WC3 strain of VP1 sequence was obtained from NCBI Protein (B2BMF7). FASTA sequence formats were obtained for all nucleic acid and peptide sequences and custom python scripts involving BioPython's Seq class were used for routine data wrangling tasks [64].
Sequence alignment
FASTA representations of each RV strain's VP1, VP3, VP4 and VP7 were aligned using JABAWS and ClustalOmega operations in a JalView environment [65-70]. Additionally, VP3-NTDs for rotavirus strains SA11 (simian), OSU (porcine), Wa (human) and all five RotaTeq reassortants (i.e., G1, G2, G3, G4, and P1) were parsed from amino acid residues 1-171 on the basis of this region's demonstrated ability to drive human MAVS protein degradation during infection by simian and porcine RV strains [8,11,12]. The VP3 NTD is highly conserved, and all 171 codons are fully represented in all 8 rotavirus strains (i.e., SA11, OSU, Wa + all five RotaTeq reassortants). A total of 28 pairwise comparisons (i.e., 8 termial, or the eighth triangular number) were performed on a per-residue basis across all conserved 171 amino acids for all eight rotaviral VP3 NTD peptide sequences to generate conservation matrices (Supplementary File 1).
Cladistics
Dendrograms were generated using the NGphylogeny.fr default pipeline [71,72], which includes documented packages: BMGE [73], FastMe [74,75], MAFFT [76], and Newick utilities [77]. Newick trees in NHX format were further visualized using iTOL software with standard parameters [78,79].
Rotavirus mRNA vaccine design
Published sequences of the Pfizer/BioNTech and Moderna mRNA vaccines for COVID-19 were obtained [80] as a vehicle for a decavalent rotavirus mRNA vaccine design entailing the five VP4 and five VP7 peptide sequences encoded across the pentavalent, live, attenuated RotaTeq vaccine cocktail [8]. To obtain open-reading frames, EMBL-EBI EMBOSS Backtranseq [81] was utilized to reverse translate all 10 peptide immunogen sequences using default parameters and a human-codon optimization algorithm. Each of the 10 total ORFs (i.e., five unique VP4 loci and five unique VP7 loci from RotaTeq reassortant G1, G2, G3, G4 and P1 strains) were then individually placed in lieu of the mRNA-1273 vaccine's SARS-CoV-2 spike ORF [8,80]. All other sequence elements of the mRNA-1273 design were retained, including the 5' UTR (i.e., an untranslated region that provides a Kozak sequence for ribosomal initiation), a start codon followed by a signal peptide sequence (i.e., providing efficient endomembrane secretion and thus granting exogenous VP4 and exogenous VP7 to the humoral response for neutralizing antibody production by activated host B-cells), three stop codons, a 3' UTR, and a polyA sequence. This decavalent (i.e., n = 10 immunogens) rotavirus mRNA cocktail would ideally be delivered using established lipid nanoparticle techniques. The complete sequence of the proposed decavalent rotavirus mRNA vaccine cocktail is provided in Supplementary File 3.
Author Contributions
Conceptualization, AB, SB, MS, JK, HD, GV; Methodology, GV; Software, GV; Validation, IE, AR, JK, HD, OD, NF, GV; Formal Analysis, AB, SB, GV; Investigation, AB, SB, GV; Resources, AB, SB, GV; Data Curation, GV; Writing — original draft preparation, AB, SB, GV; Writing — review and editing, AB, SB, MS, JK, HD, IE, AR, OD, NF, GV; Visualization, AB, SB, GV; Supervision, GV; Project Administration, GV.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding Sources
This research was not funded by any funding sources.
Acknowledgements
We thank colleagues for critical advice and key suggestions for bioinformatics and datamining approaches in the Biology Department (Dr. Matthew Gacura), the Mathematics Department (Dr. Richard Ligo), and the Forensic Investigation Center (Dr. Christopher Magno and Dr. Theodore Yeshion) at Gannon University. We are also grateful to Dr. James Warren at Penn State University-Behrend for overall manuscript guidance.
References
2. Lee KY. Rotavirus infection-associated central nervous system complications: clinicoradiological features and potential mechanisms. Clin Exp Pediatr. 2022 Oct 15;65(10):483-93.
3. Crawford SE, Ramani S, Tate JE, Parashar UD, Svensson L, Hagbom M, et al. Rotavirus infection. Nat Rev Dis Primers. 2017 Nov 9;3(1):17083.
4. Narváez CF, Mesa MC, Barreto A, Rodríguez L, Angel J. Pathogenesis of Rotavirus in Humans. In: Singh SK, editor. Human Emerging and Re‐emerging Infections. 1st ed. Wiley; 2015. p. 227-41.
5. Papa G, Borodavka A, Desselberger U. Viroplasms: Assembly and Functions of Rotavirus Replication Factories. Viruses. 2021 Jul 12;13(7):1349.
6. Caddy S, Papa G, Borodavka A, Desselberger U. Rotavirus research: 2014–2020. Virus Research. 2021 Oct;304:198499.
7. Ciarlet M, Schödel F. Development of a rotavirus vaccine: Clinical safety, immunogenicity, and efficacy of the pentavalent rotavirus vaccine, RotaTeq®. Vaccine. 2009 Dec;27:G72-81.
8. Matthijnssens J, Joelsson DB, Warakomski DJ, Zhou T, Mathis PK, Van Maanen MH, et al. Molecular and biological characterization of the 5 human-bovine rotavirus (WC3)-based reassortant strains of the pentavalent rotavirus vaccine, RotaTeq®. Virology. 2010 Aug;403(2):111-27.
9. Plosker GL. Pentavalent Rotavirus Vaccine (RotaTeq). Drugs. 2010;70(9):1165-88.
10. Van Hoek AJ, Ngama M, Ismail A, Chuma J, Cheburet S, Mutonga D, et al. A Cost Effectiveness and Capacity Analysis for the Introduction of Universal Rotavirus Vaccination in Kenya: Comparison between Rotarix and RotaTeq Vaccines. Postma M, editor. PLoS ONE. 2012 Oct 24;7(10):e47511.
11. Dai J, Agbemabiese CA, Griffin AN, Patton JT. Rotavirus capping enzyme VP3 inhibits interferon expression by inducing MAVS degradation during viral replication. Meng XJ, editor. mBio. 2023 Dec 19;14(6):e02255-23.
12. Ding S, Zhu S, Ren L, Feng N, Song Y, Ge X, et al. Rotavirus VP3 targets MAVS for degradation to inhibit type III interferon expression in intestinal epithelial cells. eLife. 2018 Nov 21;7:e39494.
13. Sharma A, Kontodimas K, Bosmann M. The MAVS Immune Recognition Pathway in Viral Infection and Sepsis. Antioxidants & Redox Signaling. 2021 Dec 1;35(16):1376-92.
14. Ren Z, Ding T, Zuo Z, Xu Z, Deng J, Wei Z. Regulation of MAVS Expression and Signaling Function in the Antiviral Innate Immune Response. Front Immunol. 2020 May 27;11:1030.
15. Missiroli S, Patergnani S, Caroccia N, Pedriali G, Perrone M, Previati M, et al. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018 Feb 28;9(3):329.
16. Vazquez C, Horner SM. MAVS Coordination of Antiviral Innate Immunity. Sullivan CS, editor. J Virol. 2015 Jul 15;89(14):6974-7.
17. Sun X, Sun L, Zhao Y, Li Y, Lin W, Chen D, et al. MAVS maintains mitochondrial homeostasis via autophagy. Cell Discov. 2016 Aug 16;2(1):16024.
18. Fu J, Hu F, Ma T, Zhao W, Tian H, Zhang Y, et al. A conventional immune regulator mitochondrial antiviral signaling protein blocks hepatic steatosis by maintaining mitochondrial homeostasis. Hepatology. 2022 Feb;75(2):403-18.
19. Wang H, Sun W, Traba J, Wu J, Qi CF, Amo L, et al. MAVS Positively Regulates Mitochondrial Integrity and Metabolic Fitness in B Cells. ImmunoHorizons. 2023 Aug 1;7(8):587-99.
20. Wang Q, Sun Z, Cao S, Lin X, Wu M, Li Y, et al. Reduced Immunity Regulator MAVS Contributes to Non-Hypertrophic Cardiac Dysfunction by Disturbing Energy Metabolism and Mitochondrial Homeostasis. Front Immunol. 2022 Jul 1;13:919038.
21. Rahman S, Copeland WC. POLG-related disorders and their neurological manifestations. Nat Rev Neurol. 2019 Jan;15(1):40-52.
22. El-Hattab AW, Scaglia F. Mitochondrial DNA Depletion Syndromes: Review and Updates of Genetic Basis, Manifestations, and Therapeutic Options. Neurotherapeutics. 2013 Apr;10(2):186-98.
23. Ropp PA, Copeland WC. Cloning and Characterization of the Human Mitochondrial DNA Polymerase, DNA Polymerase g. Genomics. 1996;36:449-58.
24. Rahman S. Mitochondrial disease in children. J Intern Med. 2020 Jun;287(6):609-33.
25. Hargreaves IP, Al Shahrani M, Wainwright L, Heales SJR. Drug-Induced Mitochondrial Toxicity. Drug Saf. 2016 Jul;39(7):661-74.
26. Finsterer J, Segall L. Drugs interfering with mitochondrial disorders. Drug and Chemical Toxicology. 2010 Apr;33(2):138-51.
27. Goodbourn S, Didcock L, Randall RE. Interferons : cell signalling, immune modulation, antiviral responses and virus countermeasures. J Gen Virol. 2000;81(10):2341-64.
28. Grandvaux N, tenOever BR, Servant MJ, Hiscott J. The interferon antiviral response: from viral invasion to evasion: Current Opinion in Infectious Diseases. 2002 Jun;15(3):259-67.
29. Rehwinkel J, Gack MU. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol. 2020 Sep;20(9):537-51.
30. Reikine S, Nguyen JB, Modis Y. Pattern Recognition and Signaling Mechanisms of RIG-I and MDA5. Front Immunol. 2014 Jul 23;5:342.
31. Barral PM, Sarkar D, Su Z zhong, Barber GN, DeSalle R, Racaniello VR, et al. Functions of the cytoplasmic RNA sensors RIG-I and MDA-5: Key regulators of innate immunity. Pharmacology & Therapeutics. 2009 Nov;124(2):219–34.
32. Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS Forms Functional Prion-like Aggregates to Activate and Propagate Antiviral Innate Immune Response. Cell. 2011 Aug;146(3):448–61.
33. Chen Y, Shi Y, Wu J, Qi N. MAVS: A Two-Sided CARD Mediating Antiviral Innate Immune Signaling and Regulating Immune Homeostasis. Front Microbiol. 2021 Sep 9;12:744348.
34. Manes NP, Nita-Lazar A. Molecular Mechanisms of the Toll-Like Receptor, STING, MAVS, Inflammasome, and Interferon Pathways. Cristea IM, editor. mSystems. 2021 Jun 29;6(3):e00336-21.
35. Samuel CE. Antiviral Actions of Interferons. Clin Microbiol Rev. 2001 Oct;14(4):778-809.
36. Bishop R. Discovery of rotavirus: Implications for Child health. J of Gastro and Hepatol. 2009 Oct;24(s3): S81-5.
37. Jiang X, Liu Y, Tan M. Histo-blood group antigens as receptors for rotavirus, new understanding on rotavirus epidemiology and vaccine strategy: Rotavirus host receptor and vaccine strategy. Emerging Microbes & Infections. 2017 Jan;6(1):1-8.
38. Ogden KM, Snyder MJ, Dennis AF, Patton JT. Predicted Structure and Domain Organization of Rotavirus Capping Enzyme and Innate Immune Antagonist VP3. Dermody TS, editor. J Virol. 2014 Aug 15;88(16):9072-85.
39. Zhang R, Jha BK, Ogden KM, Dong B, Zhao L, Elliott R, et al. Homologous 2′,5′-phosphodiesterases from disparate RNA viruses antagonize antiviral innate immunity. Proc Natl Acad Sci USA. 2013 Aug 6;110(32):13114-9.
40. Holloway G, Coulson BS. Innate cellular responses to rotavirus infection. Journal of General Virology. 2013 Jun 1;94(6):1151-60.
41. Arnold MM, Sen A, Greenberg HB, Patton JT. The Battle between Rotavirus and Its Host for Control of the Interferon Signaling Pathway. Hobman TC, editor. PLoS Pathog. 2013 Jan 24;9(1):e1003064.
42. Arnold M, Patton J. Rotavirus Antagonism of the Innate Immune Response. Viruses. 2009 Nov 24;1(3):1035-56.
43. Brand MD, Orr AL, Perevoshchikova IV, Quinlan CL. The role of mitochondrial function and cellular bioenergetics in ageing and disease: Mitochondrial function in ageing and disease. Br J Dermatol. 2013 Jul;169:1-8.
44. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. 2018 Feb;19(2):121-35.
45. Valenti D, Atlante A. Mitochondrial Bioenergetics in Different Pathophysiological Conditions 2.0. IJMS. 2022 May 16;23(10):5552.
46. Zhang Y, Qu Y, Gao K, Yang Q, Shi B, Hou P, et al. High copy number of mitochondrial DNA (mtDNA) predicts good prognosis in glioma patients. Am J Cancer Res. 2015;5(3):1207-16.
47. Betler A, Battleson S, Sawyer M, Kloecker J, Esperance I, Rahama A, et al. POLG Heterozygosity Manifests Clinically in Numerous Pediatric Cases. American Journal of Medical Research & Health Sciences. 2024;2(2):1-16.
48. Suárez-Rivero JM, Pastor-Maldonado CJ, Povea-Cabello S, Álvarez-Córdoba M, Villalón-García I, Talaverón-Rey M, et al. Mitochondria and Antibiotics: For Good or for Evil? Biomolecules. 2021 Jul 17;11(7):1050.
49. Pronicka E, Weglewska-Jurkiewicz A, Pronicki M, Sykut-Cegielska J, Kowalski P, Pajdowska M, et al. Drug-resistant epilepsia and fulminant valproate liver toxicity. Alpers-Huttenlocher syndrome in two children confirmed post mortem by identification of p.W748S mutation in POLG gene. Med Sci Monit. 2011;17(4):CR203-9.
50. Stewart JD, Horvath R, Baruffini E, Ferrero I, Bulst S, Watkins PB, et al. Polymerase γ Gene POLG determines the risk of sodium valproate-induced liver toxicity. Hepatology. 2010 Nov;52(5):1791-6.
51. Elesela S, Lukacs NW. Role of Mitochondria in Viral Infections. Life. 2021 Mar 11;11(3):232.
52. Parashar UD, Nelson EAS, Kang G. Diagnosis, management, and prevention of rotavirus gastroenteritis in children. BMJ. 2013 Dec 30;347(dec30 1):f7204-f7204.
53. Bass ES, Pappano DA, Humiston SG. Rotavirus. Pediatrics in review. 2007 May 3;28(5):183-91.
54. Sitarz KS, Elliott HR, Karaman BS, Relton C, Chinnery PF, Horvath R. Valproic acid triggers increased mitochondrial biogenesis in POLG-deficient fibroblasts. Molecular Genetics and Metabolism. 2014 May;112(1):57-63.
55. Orsucci D, Ienco EC, Siciliano G, Mancuso M. Mitochondrial disorders and drugs: what every physician should know. DIC. 2019 Jul 4;8:1-16.
56. Chan K, Truong D, Shangari N, O’Brien PJ. Drug-induced mitochondrial toxicity. Expert Opinion on Drug Metabolism & Toxicology. 2005 Dec;1(4):655-69.
57. Wong LJC, Naviaux RK, Brunetti-Pierri N, Zhang Q, Schmitt ES, Truong C, et al. Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum Mutat. 2008 Sep;29(9):E150-72.
58. Tang S, Wang J, Lee NC, Milone M, Halberg MC, Schmitt ES, et al. Mitochondrial DNA polymerase mutations: an ever expanding molecular and clinical spectrum. Journal of Medical Genetics. 2011 Oct 1;48(10):669-81.
59. Sayers EW, Bolton EE, Brister JR, Canese K, Chan J, Comeau DC, et al. Database resources of the national center for biotechnology information. Nucleic Acids Research. 2022 Jan 7;50(D1):D20-6.
60. Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res. 2016 Jan 4;44(D1):D67-72.
61. Brown GR, Hem V, Katz KS, Ovetsky M, Wallin C, Ermolaeva O, et al. Gene: a gene-centered information resource at NCBI. Nucleic Acids Research. 2015 Jan 28;43(D1):D36-42.
62. Brister JR, Ako-adjei D, Bao Y, Blinkova O. NCBI Viral Genomes Resource. Nucleic Acids Research. 2015 Jan 28;43(D1):D571-7.
63. The UniProt Consortium, Bateman A, Martin MJ, Orchard S, Magrane M, Ahmad S, et al. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Research. 2023 Jan 6;51(D1):D523-31.
64. Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, et al. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009 Jun 1;25(11):1422-3.
65. Clamp M, Cuff J, Searle SM, Barton GJ. The Jalview Java alignment editor. Bioinformatics. 2004 Feb 12;20(3):426-7.
66. Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009 May 1;25(9):1189-91.
67. Livingstone CD, Barton GJ. Protein sequence alignments: a strategy for the hierarchical analysis of residue conservation. Bioinformatics. 1993;9(6):745-56.
68. Thompson JD, Muller A, Waterhouse A, Procter J, Barton GJ, Plewniak F, et al. MACSIMS : multiple alignment of complete sequences information management system. BMC Bioinformatics. 2006 Dec;7(1):318.
69. Troshin PV, Procter JB, Sherstnev A, Barton DL, Madeira F, Barton GJ. JABAWS 2.2 distributed web services for Bioinformatics: protein disorder, conservation and RNA secondary structure. Hancock J, editor. Bioinformatics. 2018 Jun 1;34(11):1939-40.
70. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology. 2011 Jan;7(1):539.
71. Lemoine F, Domelevo Entfellner JB, Wilkinson E, Correia D, Dávila Felipe M, De Oliveira T, et al. Renewing Felsenstein’s phylogenetic bootstrap in the era of big data. Nature. 2018 Apr;556(7702):452-6.
72. Lemoine F, Correia D, Lefort V, Doppelt-Azeroual O, Mareuil F, Cohen-Boulakia S, et al. NGPhylogeny.fr: new generation phylogenetic services for non-specialists. Nucleic Acids Research. 2019 Jul 2;47(W1):W260-5.
73. Criscuolo A, Gribaldo S. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol. 2010;10(1):210.
74. Desper R, Gascuel O. Fast and Accurate Phylogeny Reconstruction Algorithms Based on the Minimum-Evolution Principle. J Comput Biol. 2002;9(5):687-705.
75. Lefort V, Desper R, Gascuel O. FastME 2.0: A Comprehensive, Accurate, and Fast Distance-Based Phylogeny Inference Program: Table 1. Mol Biol Evol. 2015 Oct;32(10):2798-800.
76. Katoh K, Standley DM. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution. 2013 Apr 1;30(4):772-80.
77. Junier T, Zdobnov EM. The Newick utilities: high-throughput phylogenetic tree processing in the U nix shell. Bioinformatics. 2010 Jul 1;26(13):1669-70.
78. Ciccarelli FD, Doerks T, Von Mering C, Creevey CJ, Snel B, Bork P. Toward Automatic Reconstruction of a Highly Resolved Tree of Life. Science. 2006 Mar 3;311(5765):1283-7.
79. Letunic I, Bork P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Research. 2021 Jul 2;49(W1):W293-6.
80. Jeong DE, McCoy M, Artiles K, Ilbay O, Fire A, Nadeau K, et al. Assemblies of putative SARS-CoV2-spike-encoding mRNA sequences for vaccines BNT-162b2 and mRNA-1273 [Internet]. 2021. Available from: https://github.com/NAalytics/Assemblies-of-putative-SARS-CoV2-spike-encoding-mRNA-sequences-for-vaccines-BNT-162b2-and-mRNA-1273/blob/21fac9b29bb8a69c1a82a8f322bb11124d65cd0b/Assemblies%20of%20putative%20SARS-CoV2-spike-encoding%20mRNA%20sequences%20for%20vaccines%20BNT-162b2%20and%20mRNA-1273.docx.pdf
81. Madeira F, Pearce M, Tivey ARN, Basutkar P, Lee J, Edbali O, et al. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Research. 2022 Jul 5;50(W1):W276-9.