Abstract
The prevalence of Non-alcoholic Fatty Liver Disease (NAFLD) is increasing, particularly in individuals who consume minimal to no alcohol. Currently, NAFLD affects at least 32% of the global population. Although not all but approximately 5-10% cases of NAFLD progress to non-alcoholic steatohepatitis (NASH), a condition arising from lipotoxicity. Patients with NASH may further develop liver fibrosis, cirrhosis, and hepatocellular carcinoma (HCC). However, it is important to note that progression from NAFLD to NASH and HCC is not inevitable for all patients. The pathogenesis of HCC in the context of NAFLD and NASH remains poorly understood, however, fat accumulation, often due to obesity or metabolic syndrome, leads to lipotoxicity. In individuals with type 2 diabetes (T2D), elevated insulin levels promote lipolysis in adipose tissue and activate lipid-synthesizing enzymes such as fatty acid synthase (FAS) and stearoyl-CoA desaturase 1 (SCD1), which contribute to lipid accumulation in the liver. Additionally, high glucose levels in T2D can alter gene regulation by activating carbohydrate response-element binding protein (ChREBP) and insulin can increase the activity of sterol regulatory element binding protein (SREBP-1c). These lipogenic transcription factors are known to directly or indirectly activate patatin-like phospholipase domain-containing protein 3 (PNPLA3). Recent research has found elevated levels of angiotensinogen (AGT) and des-angiotensin in both experimental animals and patients with NAFLD. Emerging evidence highlights the significance of genetic factors in the risk of developing NAFLD. Here we explore the association between specific genes and hepatosteatosis, detailing the roles of T2D and obesity in the progression from NAFLD to NASH, and involvement of hepatic stellate cells in liver fibrosis. Additionally, we examine the impact of genes such as PNPLA3, MBOAT7, TM6SF2, GKRP, CCN3, and AGT as well as role of single nucleotide polymorphisms in gene regulation and their contribution to the risk of NAFLD.
Keywords
Hepatosteatosis, Non-alcoholic fatty liver disease, Non-alcoholic steatohepatitis, Obesity, Type 2 diabetes mellitus
Introduction
Non-Alcoholic Fatty Liver Disease (NAFLD) is a widespread condition affecting an estimated 32.4% of the global population and playing a significant role in the onset of liver cirrhosis and hepatocellular carcinoma (HCC). Liver steatosis, or the accumulation of fat in the liver, may not invariably lead to inflammation, known as Non-Alcoholic Steatohepatitis (NASH). However, the coexistence of steatosis and inflammation can hasten the progression to liver fibrosis, more so than in NAFLD cases without inflammation. NAFLD is often linked with metabolic syndrome and Type 2 Diabetes (T2D), with one manifestation of metabolic syndrome presenting as NAFLD itself [1]. Consequently, NAFLD has been reclassified as metabolic-dysfunction associated fatty liver disease (MAFLD) and more recently as steatotic liver disease (SLD). Metabolic syndrome encompasses a range of interconnected clinical features, including insulin resistance, elevated fasting blood sugar, abnormal cholesterol levels, central obesity, and high blood pressure [2,3]. NASH is characterized by the infiltration of fat in the liver, particularly in hepatocytes, where inflammation levels increase due to macrophage activation, in the absence of significant alcohol consumption [2].
Multiple cellular interactions among liver cells, such as hepatocytes, hepatic stellate cells (HSCs), and hepatic macrophage, the Kupffer cells (KCs) occur during the development of NAFLD [4,5]. HSCs become activated by exosomes released from hepatocytes under stress from lipotoxicity, which carry various signaling molecules crucial for intercellular communication. This activation is pivotal in liver pathology leading to fibrosis by producing excessive extracellular matrix components and accelerating liver disease progression [6]. Furthermore, dysbiosis, or the imbalance of the gut microbiome, leads to increased intestinal barrier permeability allowing more bacterial endotoxins into the bloodstream. These toxins, transported to the liver via the portal vein, contribute to various health issues including liver inflammation and damage. High-fat diets, obesity, and NAFLD are associated with intestinal dysbiosis [7]. In the liver, endotoxins activate KCs, promoting the release of a myriad of pro-inflammatory cytokines, e.g., tumor necrosis factor alfa (TNFα), interleukins (IL-1, IL-6), contributing to the fibrotic transformation of the liver following hepatocyte death [8]. Further, transforming growth factor-β, which is a profibrotic mediator, activates HSCs, which in turn might contribute to the fibrotic transformation of the liver after hepatocyte death [8]. Notably, both alcoholic and non-alcoholic forms of steatohepatitis can lead to significant liver damage, highlighting the liver's remarkable regenerative ability to heal itself through a HSC-mediated repair process [9,10]. Besides, HSCs play a critical role in the development of liver fibrosis by secreting fibrin, by which a life-threatening disease, known as liver cirrhosis, might develop [11].
NAFLD is particularly common in industrialized countries, affecting 20-40% of the population. Within this group, 10-20% may develop NASH, a more severe condition often associated with metabolic syndrome [12]. The prevalence of NAFLD is alarmingly high among individuals with morbid obesity or diabetes, reaching up to 90% and 70% respectively [13,14].
Complex interactions among liver cells (hepatocytes, HSCs, and KCs) driven by overnutrition, can trigger pro-inflammatory reactions, initiating a cascade of events including inflammasome activation, hepatocyte death, and tissue regeneration, ultimately contributing to fibrogenesis [15,16]. The progression from NAFLD to NASH involves a complex interplay of various factors, including insulin resistance, and initiation of complex inflammatory reactions. Often increased levels of insulin are found among T2D individuals while the values of insulin could be lower in individuals with increased insulin clearance [17]. The transition from NAFLD to NASH involves multiple factors including insulin resistance and hypoadiponectinemia, which together with inflammation and visceral adiposity, create a pro-inflammatory state that facilitates liver damage and fibrosis. This multifactorial environment highlights the need for addressing these underlying factors for effective management and treatment of the disease. The progression of NAFLD and NASH is further illustrated by changes in gut microbiota, leading to increased TNFα and toll-like receptors (TLR4 and TLR9) agonists in the portal circulation [18,19]. Figure 1 shows the key mechanisms involved in the initiation and progression of NAFLD.

Figure 1. Key mechanisms implicated in the initiation and progression of non-alcoholic fatty liver disease (NAFLD). Adopted from one of our earlier publications on this series [197].
Insulin and Activation of Transcription Factors
Insulin interacts with the insulin receptor, a specific type of receptor known as a receptor tyrosine kinase, initiating a complex signaling cascade within cells. This interaction triggers a series of downstream effects primarily through the activation of G-protein coupled receptors. These receptors then facilitate the activation of critical signaling pathways, including the Raf serine/threonine kinases (Raf), which activates the MAPK/ERK kinase (MEK) followed by the extracellular signal-regulated kinase (ERK), (Raf/MEK/ERK), and the PI3K (phosphoinositide 3-kinase)/protein kinase B (also known as Akt), as well as phospholipase C gamma (PLCγ) pathways. These pathways are instrumental in mediating various cellular responses, including metabolism, growth, and survival of the target cells [20]. Insulin receptor induces biosynthesis of early growth response protein 1 (Egr-1) transcription factor [21] which is activated by ETS Like-1 protein (Elk-1) [22]. In addition to pancreatic β-Islets of Langerhans, Egr-1 activation also occurs in hepatocytes and lack of Egr-1 delays the hepatocyte mitotic progression [23]. Additionally, Elk-1 regulates activator protein-1 (AP-1) transcription factor [24]. Activation of Elk-1, Egr-1, and AP-1 are important in glucose homeostasis [25-27]. Egr-2 also called as Krox20, an Elk-1 related transcription factor is also activated in adipocytes, thus insulin plays an important role in the regulation of adipogenesis [28].
Insulin-mediated increased transcription of genes from upstream stimulatory factors (USF-1 and USF-2) have been observed [29]. USF binds to E-box (5’-CANNTG-3’) and can synergize with sterol regulatory element binding protein (SREBP-1c) resulting in the activation of lipogenic genes [30]. USF regulates gene transcription of fatty acid synthase (FAS). FAS converts acetyl-CoA and malonyl-CoA into palmitic acid. The FAS gene contains two E-boxes, one sterol-response element (SRE), and the liver X receptor response element (LXRE) in the proximal promoter [31]. Additional genes integral to lipogenesis, including acetyl-CoA carboxylase, ATP-citrate lyase, and mitochondrial glycerol-3-phosphate acyltransferase, also possess E-box and SRE. These genes are modulated by insulin and various nutrients, indicating a complex regulatory network that controls fat synthesis in the body. This regulation underscores the intricate interplay between genetic factors and metabolic processes, highlighting the role of specific genetic elements in the body's response to nutritional and hormonal signals [32]. Posttranslational modification by insulin which is phosphorylation and nutrients involving acetylation play a significant role in the activation of genes of fatty acid synthesis by USF [33,34].
The SREBP produces two active forms, SREBP-1a and SREBP-1c, and is involved in the regulation of lipogenic genes. It binds to SRE (5’-ATCACCCCAC-3’) or its variations. Insulin treatment causing activation of the FAS gene is mediated by SREBP-1c [35,36]. SREBP expression is regulated by LXR and glycogen synthase kinase-3 (GSK-3). c-AMP mediated phosphorylation of LXR activates SREBP-1 gene while GSK-3 phosphorylation of SREBP results in its degradation by ubiquitin ligase [37-39]. Insulin inhibits GSK-3, thus protecting SREBP-1c from its degradation, thus activating the lipogenic genes. SREBP-1c functions as a negative regulator of phosphoenolpyruvate carboxykinase (PEPCK) in the liver, the enzyme responsible for gluconeogenesis. Insulin treatment shuts down PEPCK expression, thus blocking gluconeogenesis. It occurs due to the presence of two SRE sites in the proximal promoter of PECPK [40].
SREBP-1c, which is the principal transcription factor activator, reduces fatty acid synthesis by 50%, in SREBP-1c knock-out mice indicating involvement of other transcription factor(s) [41]. The glucose influx by glucose facilitator, GLUT4 after insulin administration results into activation of carbohydrate response-element binding protein (ChREBP) which is another lipogenic transcription factor and is expressed in both hepatocytes and adipocytes [42-44]. ChREBP is a single gene and expressed by alternate promoters to produce two isoforms, ChREBPα and ChREBPβ. ChREBP heterodimerizes with Mlx and binds to 5’-CAYGNGNNNNNCNCRTG-3’ [45,46]. ChREBP is retained in cytoplasm by protein 14-3-3 and breaking of the 14-3-3- from ChREBP causes ChREBP to translocate to nucleus and mediate the transcription from carbohydrate response element (ChORE) [42]. A ChORE has been identified in acetyl-CoA carboxylase, FAS, and SCD [47]. Together with SREBP-1c, ChREBP regulates the lipogenic genes, ChREBP also regulates enzymes of glycolysis and pentose phosphate pathway, thus providing essential intermediates for lipogenesis [47,48].
Insulin mediated enhanced activity of LXR has been observed in primary hepatocytes [49] and two of LXR, LXRα and LXRβ are abundantly present in adipose tissues. LXR is a member of the nuclear receptor superfamily and heterodimerizes with the 9-cis retinoic acid receptor (RAR) and binds to 5-AGGTCANNNNAGGTCA-3’ to control insulin-mediated lipogenesis by activating FAS, ACA, and SCD genes [50]. Knock-down of LXRα and LXRα/β abolishes the expression of genes of the lipogenic processes [49,51]. LXR also activates SREBP and ChREBP, thus activating the process of lipogenesis [52,53].
Presently, there are no treatments for NAFLD or NASH except lifestyle changes which include weight reduction and less fat intake [54]. Raising the insulin sensitivity is another option for treatment which is done by peroxisome proliferator-activated receptor-γ (PPARγ) agonist; glitazones and SREBP agonist; pioglitazone which is an insulin sensitizer [55]. The ability of these drugs is manifested in reducing the levels of blood aminotransferases; alanine aminotransferase and aspartate aminotransferase (ALT,AST) which are markers of liver injuries and whose levels are increased in the event of liver injuries [56]. These drugs have been shown to decrease insulin resistance and resolve liver inflammation, but they are ineffective in resolving liver fibrosis and cirrhosis [57,58].
Role of Angiotensins Peptides in Progression of NAFLD/NASH to Fibrotic Liver
NAFLD and NASH patients which progress to fibrosis were thought to progress by increased levels of angiotensin-II (Ang-II). Ang-II is produced from precursor angiotensinogen (AGT) which is involved in wound healing and repair [59]. Ang-II binds to its receptor, type-I, and type-II, to mediate its effects. Ang-II type-I receptor blockers are common treatment currently in use for the management of hypertension. In animal models, type-I receptor blockers, such as losartan and telmisartan were observed to provide protective effects against the development of NAFLD and NASH [60,61]. In a recent study, these blockers were found to offer no benefits in resolving NAFLD and NASH [62]. The study revealed that no differences in NAFLD progression to fibrosis were observed among NAFLD patients but suggested that these blockers might prevent NAFLD development and its progression among certain individuals [62]. Correcting insulin resistance is necessary but not good enough to ameliorate NASH in most of the patients [63]. There is a need for a broader hepatoprotective drug that can resolve NAFLD/NASH and stop the process of liver fibrosis [64].
In a study, the Ang-II precursor, AGT has been implicated in liver steatosis and the development of NAFLD [65]. Using the selective expression of AGT in the liver, which is the source of circulating AGT, it was observed that liver AGT causes liver steatosis when the Western diet (WD: containing 22% w/w fat equivalent to 40 kcal% fat) is fed to mice [65]. Remarkably, overeating and lipolysis in fatty tissues release free fatty acids in the blood which infiltrate to liver causing inflammation [66]. Circulating AGT produces Ang-II which activates nuclear factor-kappaB (NF-κB) transcription factor which increases the levels of TNFα which is the one of the markers of inflammation. Increasing the levels of circulating AGT will serve as the source of increased Ang-II and source of inflammation [67].
In addition to the production of Ang II from AGT, another peptide Ang1-7 is also produced via the action of angiotensin-converting enzyme 2 (ACE2) [68]. Ang1-7 offers protective effects on liver fibrosis [69]. Ang 1-7 binds to mas-receptor and protects the liver from fibrosis by promoting collagen degradation and resolution of inflammation. The mas receptor antagonist [7-D-Ala]-Ang-(1–7) (A779) increases experimental liver injury as evidenced by TGF-β1 and hydroxyproline levels [70]. Infusion of Ang 1-7 peptide attenuated liver fibrosis as evidenced by hydroxyproline and type 1 collagen content in bile-duct ligated rat model of liver fibrosis [71]. Inhibition of HSCs was also observed as the expression of alpha smooth muscle actin (α-SMA) was reduced [71]. Inhibition of ACE with Ang 1-7 is also observed while downregulation of mas receptor is also observed with Ang 1-7. Ang 1-7 treatment decreases the expression of connective tissue growth factor (CTGF, also known as CCN2) and vascular endothelial growth factor (VEGF), these are the factors involved in fibrosis. These observations indicate that Ang 1-7 can protect the liver from fibrosis [71]. Further studies with the knock-out of ACE2 in mice establish that ACE2 is involved in the regulation of liver fibrosis [72]. Despite being a vasodilator, Ang 1-7 has failed to exhibit any vasodilatory effect in either normal or cirrhotic liver [72,73]. The vasodilatory effects of Ang 1-7 occur due to the release of nitric oxide (NO) in cirrhotic liver, the NO-mediated vasodilation is compromised due to endothelial cell dysfunction [74,75]. It is evident that Ang II and Ang1-7 are two arms of the renin-angiotensin-aldosterone system (RAAS) which possess opposite actions. For too long, ACE inhibitors as classical RAAS inhibitors have been attempted in liver diseases such as fibrosis while it is known that these inhibitors also affect the alternate pathway, i.e. Ang1-7 mediated effects. Following chronic treatments with ACE inhibitors and AT1R (Ang II type 1 receptor ) blockers, plasma renin and Ang I level rise. [76]. Chronic ACE inhibitor treatment results into increased Ang II and/or aldosterone production that is described as Ang II reactivation or aldosterone escape [77-79]. Hypothetically, activating alternate axis (Ang 1-7) in comparison to “classical axis” (Ang II) by ACE inhibitors and angiotensin II receptor blockers (ARBs) might offers protective effects for liver fibrosis [80,81].
The HSC play a pivotal role in liver fibrogenesis [82]. Ang II activates these quiescent cells and dedifferentiates into myofibroblast. Ang II promotes the release of inflammatory cytokines and the deposition of extracellular matrix (ECM). Ang II acts through the AT1R in the liver which is predominant while AT2R is also present which is comparatively less than AT1R. Gene deletion studies in mice point out that AT1R deletion protects hepatic fibrosis while AT2R deletion exacerbates fibrosis [83,84]. Animal studies defining the role of inhibitors of ACE which converts Ang I (a decapeptide produced from precursor AGT by the action of renin) into Ang II (an N-terminal octapeptide of precursor AGT, the Ang II) which is a vasopressor molecule and participates into fibrotic process after loss of hepatocytes are controversial. Treatment with losartan, an AT1R blocker failed to offer any protection against liver injury and fibrosis in NAFLD [85]. In a different study, the administration of olmesartan in a NASH model demonstrated a remarkable 70% efficacy in mitigating fibrosis [86]. Despite its widespread adoption for hypertension management, the effectiveness of AT1R (Ang II Type 1 Receptor) blockers in curbing fibrosis remains underreported, with most evidence coming from animal studies. This discrepancy in observed benefits may stem from the prolonged nature of fibrosis development in humans, coupled with the presence of only two angiotensin receptors (AT1R and AT2R) in humans, in contrast to the four receptors (AT1Ra, AT1Rb, AT2Ra, AT2Rb) identified in mice. Furthermore, the use of losartan has shown promise in diminishing liver fibrosis mediated by chronic hepatitis C virus infection with significant improvements noted after a six-month treatment regimen in patients relative to a control group [87]. In another study involving cirrhotic compensated children, 48 weeks of candesartan treatment resulted in a significant decrease of hyaluronic acid, which is a marker of fibrogenesis [88]. With no improvement in other serum markers of fibrosis, no histological data or architectural changes were reported [88]. Patients with mild fibrosis after hepatitis C infection were treated with losartan and ursodeoxycholic acid, were found to have reduced serum markers of liver fibrosis, such as transforming growth factor β1 (TGFβ1), and type IV collagen compared to only ursodeoxycholic acid. However, fibrosis scores between losartan and ursodeoxycholic vs ursodeoxycholic were not significantly different [89]. In another research investigation, the combined application of perindopril and low-dose interferon in individuals afflicted with the hepatitis C virus was shown to be effective in diminishing the levels of serum markers associated with fibrosis. These markers include hyaluronic acid, type IV collagen 7S, and procollagen III-N-peptide. However, it is important to note that histological examinations, which could provide further insight into the tissue changes, were not conducted in this study. This limitation suggests the need for future research to comprehensively assess the therapeutic impact of this treatment combination on liver fibrosis [90]. Studies using AT1R blockers on NASH patients are lacking. In a study where hypertensive patients with defined NASH were treated with losartan (50 mg/day for 48 weeks), reduction in fibrotic markers, TGF-β1, ferritin, and serum transaminases and necroinflammation were observed among five patients out of seven treated patients [91]. In another study, improvement in necroinflammation in NASH compared to NAFLD patients was observed with losartan treatment (50 mg/day for 48 weeks). The patients with NASH have less activated HSC after 48 weeks of losartan treatment [92]. In patients with advanced cirrhosis, the plasma levels of renin, Ang II, and aldosterone are increased compared with healthy individuals [93]. Chronic use of ACE inhibitors and their inhibition may not lead to sustained decreased levels of Ang II because of hyper-reninemia and possible generation of Ang II by liver chymase during ACE inhibition [94,95].
Genetic Basis of NAFLD
PNPLA3 and hepatosteatosis
NAFLD is linked to heritable components where genetic differences between individuals developing NAFLD are estimated to be 20-70% [2]. It has been found that a single nucleotide polymorphism (SNP) in the patatin-like phospholipase domain-containing protein 3 (PNPLA3) gene plays a significant role in the development of NAFLD and this SNP of PNPLA3 is a susceptibility gene of NALFD [96-98]. PNPLA3 was cloned in 2001 from mouse 3T3 preadipocytes which were feeding-inducible genes, and hence named adiponutrin [99]. It is also known as calcium-independent phospholipase A2- epsilon (IPLA2-epsilon), and chromosome 22 open reading frame20 (C22orf20) [99,100]. PNPLA family contains 9 members (PNPLA 1-9) [101]. All family members of the PNPLA family possess the patatin-like phospholipase domain (PNPLA1-9) [102]. Human PNPLA3 contains 9 exons and codes for 481 amino acids. It is localized on chromosome 22; (22q13.31) [102]. The human PNPLA3 gene is bigger (481 AA) compared to mice which is smaller (384 AAs) [101-103]. High expression of the human PNPLA3 gene has been observed in the liver while moderate expression is found in the brain, kidney, skin and adipose tissues [104]. Human PNPLA3 possesses a single nucleotide polymorphism (rs738409) which changes the amino acid, isoleucine into methionine (I148M). In a genome-wide association study (GWAS) among Hispanic, African American, and European American individuals, a strong association was observed between the 148M variant of PNPLA3 and hepatic fat levels [98]. In addition, multiple studies have linked a strong association between liver cirrhosis and the PNPLA3 148M variant [105-107]. Additionally, this variant is also associated with alcohol liver disease which proceeds first with hepatosteatosis followed by steatohepatitis [108,109]. The variant 148M is implicated in the development of fatty liver in a transgenic mouse model of NAFLD [110]. The role of PNPLA3 is controversial. The knock-out of the PNPLA3 gene in mice results in neither fatty liver nor abnormal plasma and hepatic triglyceride [111,112]. However, the knock-in of human PNPLA3/148M in mice causes hepatosteatosis after sucrose feeding, and similarly, inflammation is observed after feeding with the NASH diet [113-115].
The purified PNPLA3 protein when expressed by baculovirus in Sf9 cells has been shown to possess triacylglycerol lipase and acylglycerol transacylase activities [100]. Similarly, when Huang et al. used Sf9 purified PNPLA3 protein, observed only lipase activity against triacylglyceride, diacylglyceride, and monoacyl glyceride while transacylase activity was absent [116]. The presence of triglycerides hydrolase and retinyl esterase activities using retinyl-palmitate as substrate were observed when PNPLA3 was expressed in lower eukaryotes, such as Sacchromyces cerevisiae [117,118]. When expressed in mammalian cells, such as human embryonic kidney cells (HEK293), the purified human PNPLA3 possessed lipase activity [119]. Human PNPLA3 mutant (148M) expressed in Sf9 cells using baculovirus was shown to lose triglyceride hydrolase activity in the presence of triolein as a substrate [120].
The mechanism of action of PNPLA3 in lipid metabolism is not well understood. Protein PNPLA3 remains associated with lipid droplets in mammalian cells [120-122]. Similar to wild-type PNPLA3, the mutant 148M exhibits an abnormally increased association with lipid droplets, leading to impaired lipid metabolism and elevated lipid levels within mammalian cells. Moreover, the turnover of the mutant 148M, either through ubiquitination or autophagy-related mechanisms, is decreased compared to wild-type turnover, which remains normal across feeding and fasting cycles [123,124]. It is presumed that the activity of another homolog, PNPLA2 which is also referred to as adipose triglyceride lipase (ATGL) is inhibited because the activator protein, comparative gene identification 58 (CGI-58) also referred as abhydrolase domain containing 5 (ABHD5) can no longer competitively associate with PNPLA2 but associates at higher levels with mutant PNPLA3 [122,125,126]. PNPLA3 fails to localize to lipid droplets in CGI-58 liver knock-out cells, thus CGI-58 is needed to direct PNPLA3 association with lipid droplets [122]. There is a rare variant of PNPLA3 (rs2294918), in this variant amino acid glutamic acid (E) is changed to lysine (K) at 434 (E434K). The variant E434K PNPLA3 attenuates the I148M mediated impact on steatosis and blood enzyme levels of liver injury markers, like AST and ALT [127,128].
Regulation of the PNPLA3 Gene in T2D and Metabolic Syndrome
Regulation of the PNPLA3 gene by ChREBP and SREBP-1c are shown in Figure 2. Both ChREBP and SREBP-1c regulate PNPLA3 in human hepatocytes and in mouse liver [129-131]. SREBP-1c levels are increased in T2D because of increased levels of circulating insulin levels [132]. The binding of SREBP-1c to the promoter of PNPLA3 is increased by insulin treatment. Treatment with the inhibitor of PI3K (LY 294002) reduces the insulin-mediated promoter activity of PNPLA3 and SREBP-1c expression in HepG2 cells. The response element of SREBP-1c (SRE) is located at -97/-88 from the start site [130]. SREBP-1c cooperates with the NFY transcription factor to increase the expression of PNPLA3 [130]. Overexpression of SREBP-1c in HepG2 cells increases the expression of PNPLA3 [130]. PNPLA3 gene is regulated by ChREBP and SREBP-1c in response to hyperglycemia and increased levels of insulin which is shown in Figure 2.
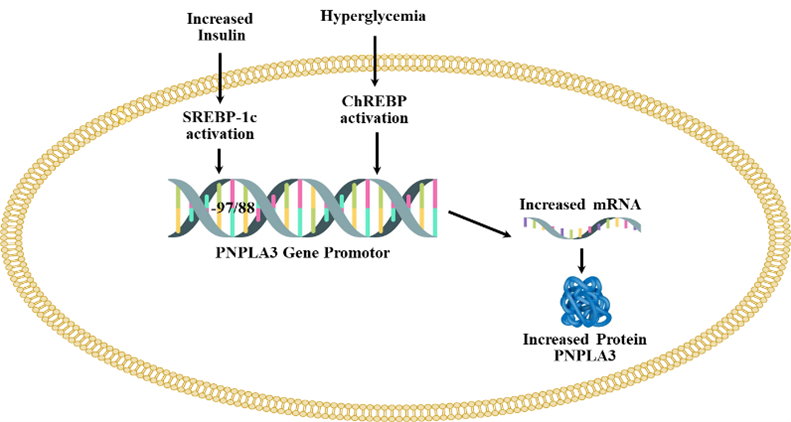
Figure 2. The upregulation of PNPLA3 by hyperglycemia and increased level of insulin. Adopted from one of our earlier publications on this series [197].
ChREBP is activated by higher blood glucose levels [133]. ChREBP transcriptional activity is regulated by intermediate products of glycolysis (glucose-6-phosphate (G-6-P), and fructose 2,6- bisphosphate), pentose phosphate pathway (xylulose-5-phosphate) [134-138], and acetylation which is a posttranscriptional modification mediated by cyclic-AMP-response element binding (CREB) protein/p300 [139]. ChREBP exists in two isoforms (ChREBPa and ChREBPb) and in normal physiological conditions, ChREBPa resides in the cytosol and upon glucose stimulation, translocates to the nucleus and induces the transcription of ChREBPb, thus linking both isoforms in the regulation of activity. ChREBPb remains localized in the nucleus which lacks the glucose inhibitory domain (LID) that is present in ChREBPa and thus ChREBPb possesses more potent transcriptional activity [140,141]. Under low glucose levels, LID-mediated inhibition of ChREBPa occurs while ChREBPb remains constitutively active [140] ChREBP is involved in de novo lipogenesis, deletion of ChREBP genes decreases liver triglycerides levels via inhibition of de novo lipogenesis [142]. Knock-down of ChREBP in ob/ob mice is shown to reduce the hepatic triglyceride levels [143]. Liver-specific silencing of ChREBP using an adenoviral-based silencing expression system reduced hepatosteatosis in ob/ob mice [144]. PNPLA3 remains in direct control of ChREBP and also SREBP-1c in both mouse and human hepatocytes. Infection of adenoviruses expressing ChREBP and SREBP-1c to mouse and human hepatocytes increases the expression of PNPLA3 [129]. Therefore, both of these transcription factors upregulate the expression of PNPLA3. Additionally, ChREBP is activated by the LXR, which forms a heterodimer with retinoid X receptors and binds to LXR response elements. Two LXR binding sites are located 2.4 kb upstream (+1) of the ChREBPα promoter, thereby inducing the expression of the ChREBP gene. It's noteworthy that ChREBPα can activate ChREBPβ, as previously mentioned [53]. The site is a response element of the nuclear hormone receptor superfamily which can also bind to thyroid receptors (TR), however, only TRb but not TRa can activate ChREBP in liver and white adipose tissues [145]. There is an indirect link of PNPLA3 mutant with the fibrogenesis of the liver. Retinoic acids (all-trans) activate RAR and retinoid X receptor (RXR) transcription factors which downregulate fibrotic genes in HSCs [146-148]. The mutant PNPLA3 (148M) decreases retinol levels causing downregulation of RAR/RXR target genes in the hepatic stellate cell line [104].
Membrane-bound O-acyltransferase domain-containing protein 7 (MBOAT7)
MBOAT7 encodes a member of membrane-bound O-acyltransferase which is integral to the membrane. The enzymatic function of the protein is not well defined, however, its transfers arachidonic acid to lysophosphatidylinositol from arachidonic-CoA, therefore known as lysophosphatidylinositol acyl-transferase 1 (LPLAT1 or LPIAT11) [149] (https://www.ncbi.nlm.nih.gov/gene/79143). MBOAT7 is involved in Land cycling in which reacylation of membrane phospholipids occurs (https://www.ncbi.nlm.nih.gov/gene/79143). Multiple variants of MBOAT7 are produced due to alternate spicing. Loss of MBOAT7 causes increased triglyceride synthesis and accumulation in hepatocytes. The substrate for triglyceride synthesis, diacylglycerol is produced due to increased turnover of phosphatidylinositol [150]. A similar decrease of MBOAT7 in stellate cells of the liver leads to an increased fibrogenic phenotype [150]. An exact opposite observation was made when hepatocyte-specific deletion of MBOAT7 was made. The cholesteryl esters were increased while no effects were observed with triglycerides [151]. Hepatic fibrosis was observed with hepatocyte-specific deficient MBOAT7 mice when treated with diet-inducing NAFLD. Initially, the polymorphism rs641738 C>T of MBOAT7 was found associated as a genetic modifier with alcohol-mediated cirrhosis and later it was linked to NAFLD and NASH [152,153]. European liver biopsy samples and Dallas heart study samples were linked to lower MBOAT protein expression and changes in the plasma phosphatidylinositol level [153]. MBOAT7 variation, rs641738 C > T was predominantly associated with Italian NAFLD and HCC patients when T was present [154]. The MBOAT7 rs641738 T allele increases an approximately 80% risk of HCC [154]. However, others did not find evidence of its association with NAFLD [155,156].
Transmembrane 6 superfamily member 2 (TM6SF2)
The function of TM6SF2 is unknown, however, studies in mice revealed that loss of TM6SF2 function causes reduced lipid export by very low-density lipoprotein (VLDL). TM6SF2 knock-out mice develop NAFLD, and hypocholesterolemia exhibiting the characteristics of human NAFLD [157]. The variant rs58542926 of TM6SF2 converts amino acid “glutamine” into lysine at amino acid position 167 when T is present and allele rs58542926 T has been associated with the increased risk of NAFLD across children and adults [158]. The loss of expression of TM6SF2 was also associated with reduced expression of PNPLA3 [157]. The variant is associated with lower levels of blood cholesterol, triglycerides and LDL-cholesterol and thus protective against cardiovascular diseases [159,160].
Glucokinase regulatory protein (GKRP)
Glucokinase is a crucial enzyme responsible for initiating glucose metabolism through glycolysis. Its activity is tightly regulated by the glucokinase regulatory protein (GKRP). Typically found in the cytoplasm, glucokinase undergoes modulation under conditions of low glucose concentration. During such times, GKRP binds to glucokinase, facilitating its translocation into the nucleus of hepatocytes. This regulatory mechanism ensures precise control over glucose utilization in hepatic cells [161]. When glucose levels rise after the meal, GKRP is released from glucokinase, thus glucokinase is transported to the cytoplasm [161]. Overexpression of GKRP, decreases fasting glucose levels and increases insulin sensitivity in diabetic mice [161]. It is proposed that GKRP increases the stability of glucokinase [161]. An SNP, rs1260326 results in the conversion of amino acid 446 from proline to leucine (P446L) in GKRP when cytosine (C) converts to thymine (T) [162]. The P446L conversion attenuates GKRP's ability to inhibit glucokinase [163]. The L446 (P446L) of GKRP has been associated with NAFLD [155,163]. Another SNP, rs780094 which is in linkage disequilibrium with rs1260326 has also been found with NAFLD and liver fibrosis severity [164-166].
Cellular communication network factor 3 (CCN3/NOV)
CCN3 is a novel adipokine that is a member of the CCN matrix protein family [167]. It is being synthesized and secreted by adipose tissue and is involved in regulating various processes, such as inflammation, organogenesis, and fibrosis [168,169]. CCN3 is encoded by the nephroblastoma overexpressing (NOV) gene [170]. Studies demonstrate association of NOV with glucose and lipid metabolism disorders, diabetes and obesity [171]. Animal studies demonstrate impairment of adipose tissues in obesity and thus increase in inflammatory molecules, including CCN3 while a decrease in adiponectin [171]. Higher serum levels of CCN3 among NAFLD patients were observed compared to controls [172]. Research indicates that CCN3 offers a protective role against apoptosis in the liver and CCN3 peptides are being attempted for treatment of NASH [173].
Adipose Tissue and Crosstalk with Liver
Adiponectin and leptin are hormones secreted by adipocytes, playing pivotal roles in metabolic regulation. Adiponectin functions to reduce liver glucose production while enhancing the utilization of glucose and fatty acids in muscle tissue. Moreover, it suppresses the secretion of inflammatory cytokines, thereby exhibiting potent anti-inflammatory properties. Reduced circulating levels of adiponectin have been associated with conditions such as NAFLD, characterized by elevated lipid accumulation in the liver [174]. Administration of adiponectin to ob/ob mice (leptin knock-out mice) results in decreased steatosis and liver inflammation [175]. The other adipocyte-derived cytokine, leptin is involved in food intake. Obese individuals have higher levels of leptin. Leptin regulates T-cell and inflammatory response [176,177]. Leptin activates stellate cells and may be involved in liver fibrosis [178]. A putative active form of adiponectin is not well defined while leptin treatment is ineffective in obese individuals. Another potent proinflammatory cytokine is resistin, its higher levels are observed among T2D patients. Resistin levels correlate with the levels of TNFα whose levels are higher among T2D patients and also NAFLDs [179,180]. TNFα plays a pivotal role in the intricate network of insulin signaling, exerting a significant influence that leads to insulin resistance. This interference underscores the complexity of metabolic regulatory mechanisms, where TNFα's interaction with insulin signaling pathways highlights its detrimental impact on glucose homeostasis. Additionally, other cytokines originating from visceral adipose tissues, including visfatin and vaspin, contribute to this regulatory landscape. Although the precise mechanisms of their actions remain partially understood, evidence suggests they play a role in reducing glucose levels. This points to a nuanced interplay between various cytokines and insulin, indicating a multifaceted regulatory environment that impacts glucose metabolism [181,182]. The cytokines from visceral fat play a significant role in NAFLD [183]. Other cytokines, such as TNFα, IL-6, and CC chemokine ligand 2 (CCL2), also known as monocyte chemoattractant protein 1 (MCP1), are also secreted by adipose tissue [181,182]. Hepatosteatosis leads to an inflammatory response whose mechanism has never been defined. In addition to adipose-derived cytokines in inflammation, liver resident Kupfer’s cell activation which is liver resident macrophage occurs which further pushes the inflammatory condition in NAFLD [184]. Chemokines also increase inflammatory cell recruitment [185].
Angiotensinogen
As mentioned above for the role of RAAS in NAFLD, the levels of AGT and des-AGT are increased in hepatosteatosis [186]. After cleavage of 10 amino acids of the N-terminal region (Ang I) of secreted AGT, the remaining portion, desAngI-AGT has been observed to possess Ang II independent function [186]. In mice lacking AGT, (Agt -/-), if desAngI-AGT is expressed using adeno-associated virus (AAV-desAngI-AGT) or AGT, caused hepatosteatosis [186]. The mechanism of AGT-mediated hepatosteaosis is unclear. It is understood that increased plasma AGT after renin action leads to increased desAngI-AGT which can mediate hepatosteatosis and so will AGT. Variation in the expression of AGT among individuals can be anticipated due to its genetic variation among humans. The human AGT gene (hAgt) possesses a number of SNP in the proximal promoter region [187]. The increased circulating levels of AGT have been associated with hypertension, although experimentally the link of these SNP with hepatosteatosis is lacking because studies were solely evaluating hypertension but not hepatosteatosis. Studies that focus on the association between AGT locus and blood pressure include linkage and genetic studies. Studies were initially demonstrated in 1992 in pairs of hypertensive siblings from American and French populations, and this study is considered the first report that SNP plays a significant role. AGT coding variants (M235T) and (T174M) [188] have been linked with the increased plasma level of AGT in hypertensive subjects when 235T and 174M were found among two independent population cohorts derived from France and Utah [189].
In addition, hAGT possesses several SNPs in the proximal promoter associated with increased gene expression and hypertension when -6A, -217A, -776T, -793A and -1074T were present, which are in linkage disequilibrium (-6A/G, -217A/G, -776T/C, -793A/G, and -1074T/G). In vitro, gene reporters have shown that haplotypes possessing -6A, -217A, -793A, and -1074T possess increased transcriptional activity [190]. In a transgenic study, Kumar et al. showed that -217A and -6A promoter variants cause increased hAGT expression in the liver, kidney and plasma [191-193]. In genetic association studies, meta-analyses of variants were conducted to investigate the correlation between the homozygous -6A and 235T alleles and hypertension [192]. Nevertheless, to date, no studies have elucidated the mechanism underlying the increased synthesis and secretion of AGT, particularly through transcription factor-mediated activation at the -6A locus [193]. One study focusing on the African-American population shows that an A/G polymorphism at -217 in the promoter of the AGT gene plays a significant role in increasing the expression of AGT and is responsible for the risk of preeclampsia, a form of hypertension occurring during pregnancy [191,194]. Other SNPs in the promoter region are still unclear and not characterized for their mechanism affecting hAGT regulation, such as -776T/C and -793 A/G. The studies from our lab demonstrate that -776T/C can bind to AP-1 transcription factors when -776C is present. Similarly, AP-1 can also bind to -793 A/G when –793A is present (unpublished data). These two sites also become the target of alcohol-mediated AGT regulation since alcohol metabolism activates the AP-1 transcription factor. The human AGT gene is an acute phase response type II secreted by the liver and regulated by inflammatory cytokines, especially IL-6, in addition to estrogen receptors (ERs), glucocorticoid receptors (GRs), and signal transducer and activator of transcription (STAT3). IL-6 activates STAT3. Activated STAT3 binds to acute phase response-II (APR II) element in the proximal promoter of AGT (-273 locus) and regulates the activation of hAGT [195]. There is one SNP present at the -1074 locus, which is variant T/G, that can bind with hepatocyte nuclear factor-3beta (HNF3β) and opens the possibility of regulation of hAGT by liver-specific orphan transcription factor(s). Increased levels of AGT have been implicated in NAFLD which is common among T2D and metabolic syndrome patients [196]. Hence, individuals harboring the -1074T variant may become susceptible to NAFLD/ALD (alcohol liver disease) following ethanol consumption due to heightened levels of IL-6-mediated AGT secretion from the liver. The genetic sequence surrounding the 1074T/G variant aligns with the consensus HNF3β sequence when T is present, whereas it exhibits two mismatches when G is present. Consequently, the genotype characterized by -6A, -217A, -776T, -793A, and -1074T may trigger enhanced AGT synthesis by the liver and elevate plasma AGT levels, thereby elucidating its implication in NAFLD. This is significant as ARBs either obstruct or mitigate the effects of Ang II in hepatosteatosis. Figure 3 represents key factors involved in the increased expression of AGT during T2D and obesity.
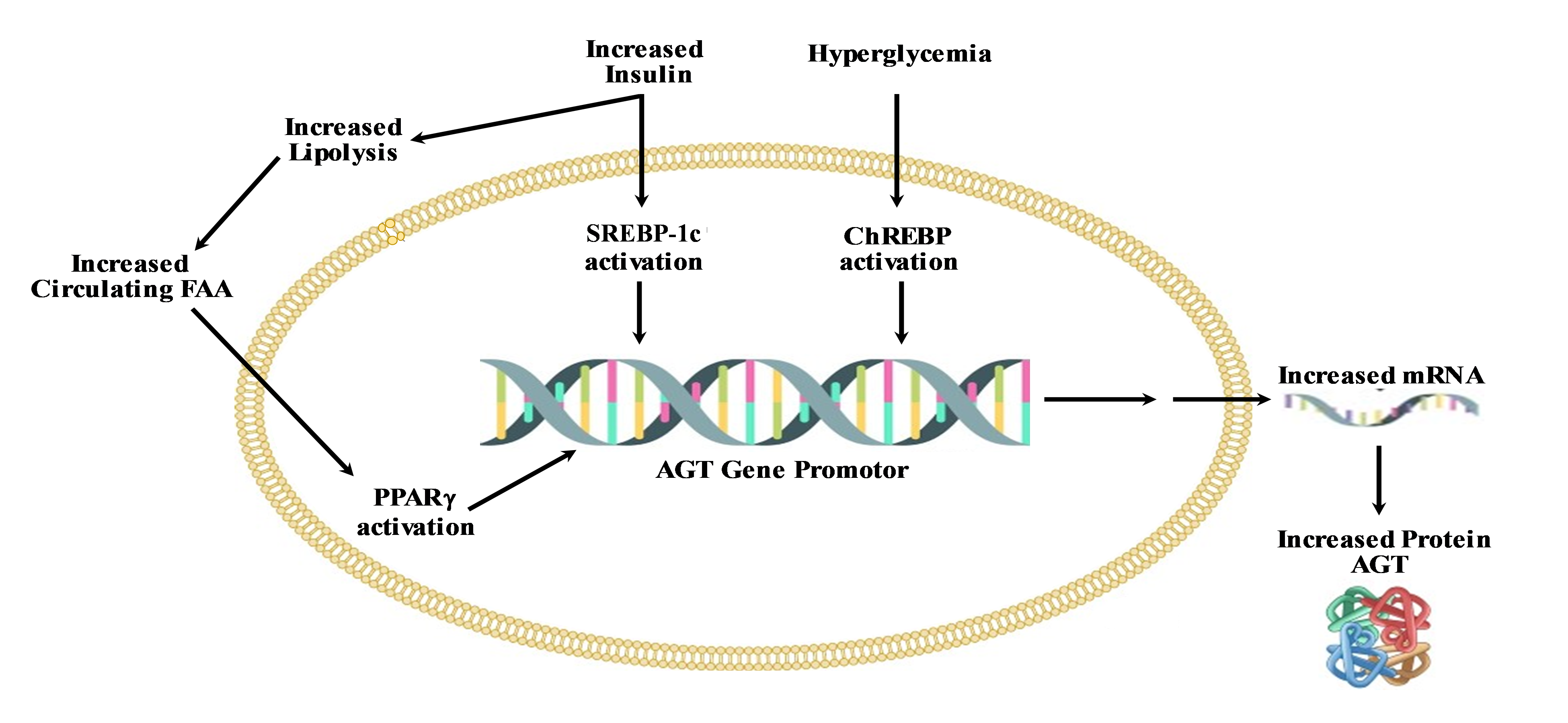
Figure 3. Key mediators of upregulation of AGT in obesity and T2D.
Conclusions
NAFLD poses a significant and escalating health challenge, not just within the United States but globally. Alarmingly, treatment expenses alone in the USA have soared to a staggering $103 billion. The severity of this issue is further accentuated by the fact that liver diseases, including cirrhosis, currently rank as the 12th leading cause of mortality in the USA, according to data from 2014. The surge in NAFLD prevalence can be attributed to the escalating incidence of obesity and metabolic syndrome over the past two decades. These interlinked conditions have substantially contributed to the rise in cases of fatty liver disease, with NAFLD prevalence ranging from 30% to 90% among individuals with obesity and approximately 70% in those with T2D. Presently, diagnosing NAFLD or its more severe manifestation, NASH, relies on a multifaceted assessment of clinical parameters rather than a singular definitive marker. However, despite the mounting prevalence and associated economic burdens, currently there are no pharmaceutical interventions specifically endorsed for the treatment of either NAFLD or NASH. Effectively tackling NAFLD and NASH demands a comprehensive understanding of the intricate molecular mechanisms governing the initiation and progression of hepatic steatosis. Pinpointing these pivotal events holds the potential to pave the way for targeted therapeutic interventions aimed at arresting or even reversing the disease trajectory. Such advancements offer a glimmer of hope for the millions affected by these debilitating conditions, promising improved outcomes and quality of life.
References
2. Powell EE, Wong VW, Rinella M. Non-alcoholic fatty liver disease. The Lancet. 2021 Jun 5;397(10290):2212-24.
3. Huang PL. A comprehensive definition for metabolic syndrome. Disease Models & Mechanisms. 2009 Apr 30;2(5-6):231-7.
4. Duarte N, Coelho IC, Patarrão RS, Almeida JI, Penha-Gonçalves C, Macedo MP. How Inflammation Impinges on NAFLD: A Role for Kupffer Cells. Biomed Res Int. 2015;2015:984578.
5. Kazankov K, Jørgensen SM, Thomsen KL, Møller HJ, Vilstrup H, George J, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nature Reviews Gastroenterology & Hepatology. 2019 Mar;16(3):145-59.
6. Luo X, Luo SZ, Xu ZX, Zhou C, Li ZH, Zhou XY, et al. Lipotoxic hepatocyte-derived exosomal miR-1297 promotes hepatic stellate cell activation through the PTEN signaling pathway in metabolic-associated fatty liver disease. World J Gastroenterol. 2021 Apr 14;27(14):1419-1434.
7. Fianchi F, Liguori A, Gasbarrini A, Grieco A, Miele L. Nonalcoholic fatty liver disease (NAFLD) as model of gut–liver axis interaction: From pathophysiology to potential target of treatment for personalized therapy. International Journal of Molecular Sciences. 2021 Jun 17;22(12):6485.
8. Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. 2010 Aug;30(3):245-57.
9. Maher JJ. Alcoholic steatosis and steatohepatitis. Seminars in Gastrointestinal Disease. 2002 Jan 1;13(1):31-9.
10. Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiological Reviews. 2010 Jul;90(3):1165-94.
11. Geh D, Anstee QM, Reeves HL. NAFLD-associated HCC: progress and opportunities. Journal of Hepatocellular Carcinoma. 2021 Apr 8:223-39.
12. Mitra S, De A, Chowdhury A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl Gastroenterol Hepatol. 2020 Apr 5;5:16.
13. Dai W, Ye L, Liu A, Wen SW, Deng J, Wu X, et al. Prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus: a meta-analysis. Medicine. 2017 Sep 1;96(39):e8179.
14. Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nature Reviews Gastroenterology & Hepatology. 2018 Jan;15(1):11-20.
15. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nature Medicine. 2018 Jul;24(7):908-22.
16. Sanyal AJ. Past, present and future perspectives in nonalcoholic fatty liver disease. Nature Reviews Gastroenterology & Hepatology. 2019 Jun;16(6):377-86.
17. Sugiyama S, Jinnouchi H, Hieshima K, Kurinami N, Jinnouchi K, Yoshida A, et al. Potential identification of type 2 diabetes with elevated insulin clearance. NEJM Evidence. 2022 Mar 22;1(4):EVIDoa2100052.
18. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. 2016 Aug 1;65(8):1038-48.
19. Khanmohammadi S, Kuchay MS. Toll-like receptors and metabolic (dysfunction)-associated fatty liver disease. Pharmacological Research. 2022 Nov 1;185:106507.
20. Thiel G, Guethlein LA, Rössler OG. Insulin-responsive transcription factors. Biomolecules. 2021 Dec 15;11(12):1886.
21. Harada S, Smith RM, Smith JA, White MF, Jarett L. Insulin-induced egr-1 and c-fos expression in 32D cells requires insulin receptor, Shc, and mitogen-activated protein kinase, but not insulin receptor substrate-1 and phosphatidylinositol 3-kinase activation. Journal of Biological Chemistry. 1996 Nov 22;271(47):30222-6.
22. Thiel G, Backes TM, Guethlein LA, Rössler OG. Critical Protein–Protein Interactions Determine the Biological Activity of Elk-1, a Master Regulator of Stimulus-Induced Gene Transcription. Molecules. 2021 Oct 11;26(20):6125.
23. Liao Y, Shikapwashya ON, Shteyer E, Dieckgraefe BK, Hruz PW, Rudnick DA. Delayed hepatocellular mitotic progression and impaired liver regeneration in early growth response-1-deficient mice. Journal of Biological Chemistry. 2004 Oct 8;279(41):43107-16.
24. Müller I, Endo T, Thiel G. Regulation of AP‐1 activity in glucose‐stimulated insulinoma cells. Journal of Cellular Biochemistry. 2010 Aug 15;110(6):1481-94.
25. Müller I, Rössler OG, Wittig C, Menger MD, Thiel G. Critical role of Egr transcription factors in regulating insulin biosynthesis, blood glucose homeostasis, and islet size. Endocrinology. 2012 Jul 1;153(7):3040-53.
26. Lesch A, Backes TM, Langfermann DS, Rössler OG, Laschke MW, Thiel G. Ternary complex factor regulates pancreatic islet size and blood glucose homeostasis in transgenic mice. Pharmacological Research. 2020 Sep 1;159:104983.
27. Backes TM, Langfermann DS, Lesch A, Rössler OG, Laschke MW, Vinson C, et al. Regulation and function of AP-1 in insulinoma cells and pancreatic β-cells. Biochemical Pharmacology. 2021 Nov 1;193:114748.
28. Wang W, Huang L, Huang Y, Yin JW, Berk AJ, Friedman JM, et al. Mediator MED23 links insulin signaling to the adipogenesis transcription cascade. Developmental Cell. 2009 May 19;16(5):764-71.
29. Ferre‐D'Amare AR, Pognonec P, Roeder RG, Burley SK. Structure and function of the b/HLH/Z domain of USF. The EMBO Journal. 1994 Jan 1;13(1):180-9.
30. Griffin MJ, Wong RH, Pandya N, Sul HS. Direct interaction between USF and SREBP-1c mediates synergistic activation of the fatty-acid synthase promoter. Journal of Biological Chemistry. 2007 Feb 23;282(8):5453-67.
31. Latasa MJ, Griffin MJ, Moon YS, Kang C, Sul HS. Occupancy and function of the− 150 sterol regulatory element and− 65 E-box in nutritional regulation of the fatty acid synthase gene in living animals. Molecular and Cellular Biology. 2003 Aug 1;23(16):5896-907.
32. Wong RH, Sul HS. Insulin signaling in fatty acid and fat synthesis: a transcriptional perspective. Current Opinion in Pharmacology. 2010 Dec 1;10(6):684-91.
33. Wong RH, Chang I, Hudak CS, Hyun S, Kwan HY, Sul HS. A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell. 2009 Mar 20;136(6):1056-72.
34. Wang Y, Viscarra J, Kim SJ, Sul HS. Transcriptional regulation of hepatic lipogenesis. Nature reviews Molecular Cell Biology. 2015 Nov;16(11):678-89.
35. Dorotea D, Koya D, Ha H. Recent Insights Into SREBP as a Direct Mediator of Kidney Fibrosis via Lipid-Independent Pathways. Front Pharmacol. 2020 Mar 17;11:265.
36. Foretz M, Pacot C, Dugail I, Lemarchand P, Guichard C, Le Lièpvre X, et al. ADD1/SREBP-1c is required in the activation of hepatic lipogenic gene expression by glucose. Molecular and Cellular Biology. 1999 May 1;19(5):3760-8.
37. Bengoechea-Alonso MT, Ericsson J. A phosphorylation cascade controls the degradation of active SREBP1. Journal of Biological Chemistry. 2009 Feb 27;284(9):5885-95.
38. Lu M, Shyy JY. Sterol regulatory element-binding protein 1 is negatively modulated by PKA phosphorylation. American Journal of Physiology-Cell Physiology. 2006 Jun;290(6):C1477-86.
39. Yamamoto T, Shimano H, Inoue N, Nakagawa Y, Matsuzaka T, Takahashi A, et al. Protein kinase A suppresses sterol regulatory element-binding protein-1C expression via phosphorylation of liver X receptor in the liver. Journal of Biological Chemistry. 2007 Apr 20;282(16):11687-95.
40. Chakravarty K, Wu SY, Chiang CM, Samols D, Hanson RW. SREBP-1c and Sp1 interact to regulate transcription of the gene for phosphoenolpyruvate carboxykinase (GTP) in the liver. Journal of Biological Chemistry. 2004 Apr 9;279(15):15385-95.
41. Liang G, Yang J, Horton JD, Hammer RE, Goldstein JL, Brown MS. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. Journal of Biological Chemistry. 2002 Mar 15;277(11):9520-8.
42. Abdul-Wahed A, Guilmeau S, Postic C. Sweet sixteenth for ChREBP: established roles and future goals. Cell Metabolism. 2017 Aug 1;26(2):324-41.
43. Filhoulaud G, Guilmeau S, Dentin R, Girard J, Postic C. Novel insights into ChREBP regulation and function. Trends in Endocrinology & Metabolism. 2013 May 1;24(5):257-68.
44. Postic C, Dentin R, Denechaud PD, Girard J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu. Rev. Nutr.. 2007 Aug 21;27:179-92.
45. Ma L, Robinson LN, Towle HC. ChREBP• Mlx is the principal mediator of glucose-induced gene expression in the liver. Journal of Biological Chemistry. 2006 Sep 29;281(39):28721-30.
46. Ma L, Tsatsos NG, Towle HC. Direct role of ChREBP· Mlx in regulating hepatic glucose-responsive genes. Journal of Biological Chemistry. 2005 Mar 25;280(12):12019-27.
47. Ishii S, IIzuka K, Miller BC, Uyeda K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proceedings of the National Academy of Sciences. 2004 Nov 2;101(44):15597-602.
48. Baraille F, Planchais J, Dentin R, Guilmeau S, Postic C. Integration of ChREBP-mediated glucose sensing into whole body metabolism. Physiology. 2015 Nov;30(6):428-37.
49. Tobin KA, Ulven SM, Schuster GU, Steineger HH, Andresen SM, Gustafsson JÅ, et al. Liver X receptors as insulin-mediating factors in fatty acid and cholesterol biosynthesis. Journal of Biological Chemistry. 2002 Mar 22;277(12):10691-7.
50. Wang B, Tontonoz P. Liver X receptors in lipid signalling and membrane homeostasis. Nature Reviews Endocrinology. 2018 Aug;14(8):452-63.
51. Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, et al. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXRα. Cell. 1998 May 29;93(5):693-704.
52. Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRα and LXRβ. Genes & Development. 2000 Nov 15;14(22):2819-30.
53. Cha JY, Repa JJ. The liver X receptor (LXR) and hepatic lipogenesis: the carbohydrate-response element-binding protein is a target gene of LXR. Journal of Biological Chemistry. 2007 Jan 5;282(1):743-51.
54. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018 Jan 1;67(1):328-57.
55. Khan RS, Bril F, Cusi K, Newsome PN. Modulation of insulin resistance in nonalcoholic fatty liver disease. Hepatology. 2019 Aug;70(2):711-24.
56. Sookoian S, Castaño GO, Scian R, Gianotti TF, Dopazo H, Rohr C, et al. Serum aminotransferases in nonalcoholic fatty liver disease are a signature of liver metabolic perturbations at the amino acid and Krebs cycle level. The American Journal of Clinical Nutrition. 2016 Feb 1;103(2):422-34.
57. Desouza CV, Shivaswamy V. Pioglitazone in the treatment of type 2 diabetes: safety and efficacy review. Clinical Medicine Insights. Endocrinology and Diabetes. 2010 Aug 3;3:43-51.
58. Kendall DM. Thiazolidinediones: The case for early use. Diabetes Care. 2006 Jan 1;29(1):154-7.
59. Morris EM, Fletcher JA, Thyfault JP, Rector RS. The role of angiotensin II in nonalcoholic steatohepatitis. Molecular and Cellular Endocrinology. 2013 Sep 25;378(1-2):29-40.
60. Borém LM, Neto JF, Brandi IV, Lelis DF, Santos SH. The role of the angiotensin II type I receptor blocker telmisartan in the treatment of non-alcoholic fatty liver disease: a brief review. Hypertension Research. 2018 Jun;41(6):394-405.
61. Yoshiji H, Noguchi R, Ikenaka Y, Namisaki T, Kitade M, Kaji K, et al. Losartan, an angiotensin-II type 1 receptor blocker, attenuates the liver fibrosis development of non-alcoholic steatohepatitis in the rat. BMC Research Notes. 2009;2:70.
62. Kim KM, Roh JH, Lee S, Yoon JH. Clinical implications of renin–angiotensin system inhibitors for development and progression of non-alcoholic fatty liver disease. Scientific Reports. 2021 Feb 3;11(1):2884.
63. Lonardo A, Bellentani S, Ratziu V, Loria P. Insulin resistance in nonalcoholic steatohepatitis: necessary but not sufficient–death of a dogma from analysis of therapeutic studies?. Expert Review of Gastroenterology & Hepatology. 2011 Apr 1;5(2):279-89.
64. Mayo Clinic. Nonalcoholic fatty liver disease. 2021.
65. Tao XR, Rong JB, Lu HS, Daugherty A, Shi P, Ke CL, et al. Angiotensinogen in hepatocytes contributes to Western diet-induced liver steatosis. Journal of Lipid Research. 2019 Dec 1;60(12):1983-95.
66. Perdomo CM, Frühbeck G, Escalada J. Impact of nutritional changes on nonalcoholic fatty liver disease. Nutrients. 2019 Mar 21;11(3):677.
67. Ji Y, Wang Z, Li Z, Zhang A, Jin Y, Chen H, et al. Angiotensin II enhances proliferation and inflammation through AT1/PKC/NF-κB signaling pathway in hepatocellular carcinoma cells. Cellular Physiology and Biochemistry. 2016 Jul 1;39(1):13-32.
68. Jiang F, Yang J, Zhang Y, Dong M, Wang S, Zhang Q, et al. Angiotensin-converting enzyme 2 and angiotensin 1–7: novel therapeutic targets. Nature Reviews Cardiology. 2014 Jul;11(7):413-26.
69. Cai SM, Yang RQ, Li Y, Ning ZW, Zhang LL, Zhou GS, et al. Angiotensin-(1–7) improves liver fibrosis by regulating the NLRP3 inflammasome via redox balance modulation. Antioxidants & Redox Signaling. 2016 May 10;24(14):795-812.
70. Pereira RM, Dos Santos RA, Teixeira MM, Leite VH, Costa LP, da Costa Dias FL, et al. The renin–angiotensin system in a rat model of hepatic fibrosis: Evidence for a protective role of Angiotensin-(1–7). Journal of Hepatology. 2007 Apr 1;46(4):674-81.
71. Lubel JS, Herath CB, Jia Z, Burrell LM, Angus PW. Angiotensin 1-7 reduces bile duct proliferation and hepatic fibrosis in the bile duct ligated rat. Hepatology. 2007 Oct 1;46(4):706A-707A.
72. Oeterreicher CH, Seki E, De Minicis S, Taura K, Penz-Oesterreicher M, Kluwe J, et al. Angiotensin-converting-enzyme 2 is a negative regulator of chronic liver injury. Hepatology. 2007 Oct 1;46(4):298A-299A.
73. Lubel JS, Herath CB, Warner FJ, Jia ZY, Burrell LM, Angus PW. Upregulation of the ACE2/ANG1-7/MAS receptor axis in the bile duct ligation (BDL) model of hepatic fibrosis does not affect hepatic sinusoidal resistance. Journal of Gastroenterology and Hepatology. 2006 Oct 1;21:A332.
74. Hendrickson H, Chatterjee S, Cao S, Ruiz MM, Sessa WC, Shah V. Influence of caveolin on constitutively activated recombinant eNOS: insights into eNOS dysfunction in BDL rat liver. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2003 Sep;285(3):G652-60.
75. Iwakiri Y, Groszmann RJ. Vascular endothelial dysfunction in cirrhosis. Journal of hepatology. 2007 May 1;46(5):927-34.
76. Mooser V, Nussberger J, Juillerat L, Burnier M, Waeber B, Bidiville J, et al. Reactive hyperreninemia is a major determinant of plasma angiotensin II during ACE inhibition. Journal of cardiovascular pharmacology. 1990 Feb 1;15(2):276-82.
77. MacFadyen RJ, Lee AF, Morton JJ, Pringle SD, Struthers AD. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure?. Heart. 1999 Jul 1;82(1):57-61.
78. Sharman DC, Morris AD, Struthers AD. Gradual reactivation of vascular angiotensin I to angiotensin II conversion during chronic ACE inhibitor therapy in patients with diabetes mellitus. Diabetologia. 2007 Oct;50:2061-6.
79. van de Wal RM, Plokker HW, Lok DJ, Boomsma F, van der Horst FA, van Veldhuisen DJ, et al. Determinants of increased angiotensin II levels in severe chronic heart failure patients despite ACE inhibition. International journal of cardiology. 2006 Jan 26;106(3):367-72.
80. Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, Diz DI, Gallagher PE. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005 May 24;111(20):2605-10.
81. Ishiyama Y, Gallagher PE, Averill DB, Tallant EA, Brosnihan KB, Ferrario CM. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension. 2004 May 1;43(5):970-6.
82. Bataller R, Brenner DA. Hepatic stellate cells as a target for the treatment of liver fibrosis. Semin Liver Dis. 2001 Aug;21(3):437-51.
83. Kanno K, Tazuma S, Chayama K. AT1A-deficient mice show less severe progression of liver fibrosis induced by CCl4. Biochemical and Biophysical Research Communications. 2003 Aug 15;308(1):177-83.
84. Nabeshima Y, Tazuma S, Kanno K, Hyogo H, Iwai M, Horiuchi M, et al. Anti-fibrogenic function of angiotensin II type 2 receptor in CCl4-induced liver fibrosis. Biochemical and Biophysical Research Communications. 2006 Aug 4;346(3):658-64.
85. Ibañez P, Solis N, Pizarro M, Aguayo G, Duarte I, Miquel JF, et al. Effect of losartan on early liver fibrosis development in a rat model of nonalcoholic steatohepatitis. Journal of Gastroenterology and Hepatology. 2007 Jun;22(6):846-51.
86. Hirose A, Ono M, Saibara T, Nozaki Y, Masuda K, Yoshioka A, et al. Angiotensin II type 1 receptor blocker inhibits fibrosis in rat nonalcoholic steatohepatitis. Hepatology. 2007 Jun;45(6):1375-81.
87. Sookoian S, Fernández MA, Castaño G. Effects of six months losartan administration on liver fibrosis in chronic hepatitis C patients: A pilot study. World J Gastroenterol. 2005;11(48):7560-3.
88. Debernardi-Venon W, Martini S, Biasi F, Vizio B, Termine A, Poli G, et al. AT1 receptor antagonist Candesartan in selected cirrhotic patients: effect on portal pressure and liver fibrosis markers. Journal of Hepatology. 2007 Jun 1;46(6):1026-33.
89. Terui Y, Saito T, Watanabe H, Togashi H, Kawata S, Kamada Y, et al. Effect of angiotensin receptor antagonist on liver fibrosis in early stages of chronic hepatitis C. Hepatology. 2002 Oct 1;36(4):1022.
90. Yoshiji H, Noguchi R, Fukui H. Combined effect of an ACE inhibitor, perindopril, and interferon on liver fibrosis markers in patients with chronic hepatitis C. Journal of Gastroenterology. 2005 Feb 1;40(2):215-6.
91. Yokohama S, Yoneda M, Haneda M, Okamoto S, Okada M, Aso K, et al. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology. 2004 Nov 1;40(5):1222-5.
92. Yokohama S, Tokusashi Y, Nakamura K, Tamaki Y, Okamoto S, Okada M, et al. Inhibitory effect of angiotensin II receptor antagonist on hepatic stellate cell activation in non-alcoholic steatohepatitis. World J Gastroenterol. 2006 Jan 14;12(2):322-6.
93. Zhang Z, Feng H, Leng X, Ma F, Wang B, Du R. The levels of renin activity, angiotensin converting enzyme and angiotensin II in cirrhotic patients with ascites undergoing portacaval shunt. Zhonghua wai ke za zhi [Chinese Journal of Surgery]. 1999 Jun 1;37(6):366-8.
94. Komeda K, Jin D, Takai S, Hayashi M, Takeshita A, Shibayama Yet al. Significance of chymase‐dependent angiotensin II formation in the progression of human liver fibrosis. Hepatology Research. 2008 May;38(5):501-10.
95. Shimizu S, Satomura K, Aramaki T, Katsuta Y, Takano T, Omoto Y. Hepatic chymase level in chronic hepatitis: co-localization of chymase with fibrosis. Hepatology research. 2003 Sep 1;27(1):62-6.
96. Eslam M, George J. Genetic contributions to NAFLD: leveraging shared genetics to uncover systems biology. Nature reviews Gastroenterology & hepatology. 2020 Jan;17(1):40-52.
97. Meffert PJ, Repp KD, Völzke H, Weiss FU, Homuth G, Kühn JP, Lerch MM, Aghdassi AA. The PNPLA3 SNP rs738409: G allele is associated with increased liver disease-associated mortality but reduced overall mortality in a population-based cohort. Journal of Hepatology. 2018 Apr 1;68(4):858-60.
98. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nature Genetics. 2008 Dec;40(12):1461-5.
99. Baulande S, Lasnier F, Lucas M, Pairault J. Adiponutrin, a transmembrane protein corresponding to a novel dietary-and obesity-linked mRNA specifically expressed in the adipose lineage. Journal of Biological Chemistry. 2001 Sep 7;276(36):33336-44.
100. Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. Journal of Biological Chemistry. 2004 Nov 19;279(47):48968-75.
101. Wilson PA, Gardner SD, Lambie NM, Commans SA, Crowther DJ. Characterization of the human patatin-like phospholipase familys⃞. Journal of Lipid Research. 2006 Sep 1;47(9):1940-9.
102. Dong XC. PNPLA3—a potential therapeutic target for personalized treatment of chronic liver disease. Frontiers in Medicine. 2019 Dec 17;6:304.
103. Huang Y, He S, Li JZ, Seo YK, Osborne TF, Cohen JC et al. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proceedings of the National Academy of Sciences. 2010 Apr 27;107(17):7892-7.
104. Bruschi FV, Claudel T, Tardelli M, Caligiuri A, Stulnig TM, Marra F, et al. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology. 2017 Jun;65(6):1875-90.
105. Falleti E, Fabris C, Cmet S, Cussigh A, Bitetto D, Fontanini E, et al. PNPLA3 rs738409C/G polymorphism in cirrhosis: relationship with the aetiology of liver disease and hepatocellular carcinoma occurrence. Liver International. 2011 Sep;31(8):1137-43.
106. Kupcinskas J, Valantiene I, Varkalaitė G, Steponaitiene R, Skieceviciene J, Sumskiene J, et al. PNPLA3 and RNF7 Gene Variants are Associated with the Risk of Developing Liver Fibrosis and Cirrhosis in an Eastern European Population. Journal of Gastrointestinal & Liver Diseases. 2017 Mar;26(1):37-43.
107. Shen JH, Li YL, Li D, Wang NN, Jing L, Huang YH. The rs738409 (I148M) variant of the PNPLA3 gene and cirrhosis: a meta-analysis. Journal of Lipid Research. 2015 Jan 1;56(1):167-75.
108. Burza MA, Molinaro A, Attilia ML, Rotondo C, Attilia F, Ceccanti M, et al. PNPLA3 I148M (rs738409) genetic variant and age at onset of at‐risk alcohol consumption are independent risk factors for alcoholic cirrhosis. Liver International. 2014 Apr;34(4):514-20.
109. Chamorro AJ, Torres JL, Mirón‐Canelo JA, González‐Sarmiento R, Laso FJ, Marcos M. Systematic review with meta‐analysis: the I148M variant of patatin‐like phospholipase domain‐containing 3 gene (PNPLA3) is significantly associated with alcoholic liver cirrhosis. Alimentary pharmacology & therapeutics. 2014 Sep;40(6):571-81.
110. Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, Castro-Perez J, Cohen JC, et al. Chronic overexpression of PNPLA3 I148M in mouse liver causes hepatic steatosis. The Journal of Clinical Investigation. 2012 Nov 1;122(11):4130-44.
111. Basantani MK, Sitnick MT, Cai L, Brenner DS, Gardner NP, Li JZ, et al. Pnpla3/Adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome [S]. Journal of Lipid Research. 2011 Feb 1;52(2):318-29.
112. Chen W, Chang B, Li L, Chan L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology. 2010 Sep;52(3):1134-42. doi: 10.1002/hep.23812. Erratum in: Hepatology. 2010 Dec;52(6):2250.
113. BasuRay S, Smagris E, Cohen JC, Hobbs HH. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology. 2017 Oct;66(4):1111-24.
114. Lindén D, Ahnmark A, Pingitore P, Ciociola E, Ahlstedt I, Andréasson AC, et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Molecular Metabolism. 2019 Apr 1;22:49-61.
115. Smagris E, BasuRay S, Li J, Huang Y, Lai KM, Gromada J, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology. 2015 Jan;61(1):108-18.
116. Huang Y, Cohen JC, Hobbs HH. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. Journal of Biological Chemistry. 2011 Oct 28;286(43):37085-93.
117. Pingitore P, Pirazzi C, Mancina RM, Motta BM, Indiveri C, Pujia A, et al. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids. 2014 Apr 1;1841(4):574-80.
118. Pirazzi C, Valenti L, Motta BM, Pingitore P, Hedfalk K, Mancina RM, et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Human Molecular Genetics. 2014 Aug 1;23(15):4077-85.
119. Lake AC, Sun Y, Li JL, Kim JE, Johnson JW, Li D, et al. Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. Journal of Lipid Research. 2005 Nov 1;46(11):2477-87.
120. He S. A Sequence Variation (I148M) in PNPLA3 Associated with Nonalcoholic Fatty Liver Disease Disrupts Triglyceride Hydrolysis. J Biol Chem 2010 Feb 26;285(9):6706-15.
121. Murugesan S, Goldberg EB, Dou E, Brown WJ. Identification of diverse lipid droplet targeting motifs in the PNPLA family of triglyceride lipases. PLoS One. 2013 May 31;8(5):e64950.
122. Wang Y, Kory N, Basu Ray S, Cohen JC, Hobbs HH. PNPLA3, CGI‐58, and inhibition of hepatic triglyceride hydrolysis in mice. Hepatology. 2019 Jun;69(6):2427-41.
123. BasuRay S, Wang Y, Smagris E, Cohen JC, Hobbs HH. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proceedings of the National Academy of Sciences. 2019 May 7;116(19):9521-6.
124. Negoita F, Blomdahl J, Wasserstrom S, Winberg ME, Osmark P, Larsson S, et al. PNPLA3 variant M148 causes resistance to starvation‐mediated lipid droplet autophagy in human hepatocytes. Journal of Cellular Biochemistry. 2019 Jan;120(1):343-56.
125. Chamoun Z, Vacca F, Parton RG, Gruenberg J. PNPLA3/adiponutrin functions in lipid droplet formation. Biology of the Cell. 2013 May;105(5):219-33.
126. Yang A, Mottillo EP, Mladenovic-Lucas L, Zhou L, Granneman JG. Dynamic interactions of ABHD5 with PNPLA3 regulate triacylglycerol metabolism in brown adipocytes. Nature Metabolism. 2019 May;1(5):560-9.
127. Donati B, Motta BM, Pingitore P, Meroni M, Pietrelli A, Alisi A, et al. The rs2294918 E434K variant modulates patatin‐like phospholipase domain‐containing 3 expression and liver damage. Hepatology. 2016 Mar;63(3):787-98.
128. Schwartz BE, Rajagopal V, Smith C, Cohick E, Whissell G, Gamboa M, et al. Discovery and targeting of the signaling controls of PNPLA3 to effectively reduce transcription, expression, and function in pre-clinical NAFLD/NASH settings. Cells. 2020 Oct 7;9(10):2247.
129. Dubuquoy C, Robichon C, Lasnier F, Langlois C, Dugail I, Foufelle F, et al. Distinct regulation of adiponutrin/PNPLA3 gene expression by the transcription factors ChREBP and SREBP1c in mouse and human hepatocytes. Journal of Hepatology. 2011 Jul 1;55(1):145-53.
130. Liang H, Xu J, Xu F, Liu H, Yuan D, Yuan S, et al. The SRE Motif in the Human PNPLA3 Promoter (− 97 to− 88 bp) Mediates Transactivational Effects of SREBP‐1c. Journal of Cellular Physiology. 2015 Sep;230(9):2224-32.
131. Perttilä J, Huaman-Samanez C, Caron S, Tanhuanpää K, Staels B, Yki-Järvinen H, et al. PNPLA3 is regulated by glucose in human hepatocytes, and its I148M mutant slows down triglyceride hydrolysis. American Journal of Physiology-Endocrinology and Metabolism. 2012 May 1;302(9):E1063-9.
132. Ferre P, Foufelle F. SREBP-1c transcription factor and lipid homeostasis: clinical perspective. Hormone Research. 2007 Jul 1;68(2):72-82.
133. Li MV, Chen W, Poungvarin N, Imamura M, Chan L. Glucose-mediated transactivation of carbohydrate response element-binding protein requires cooperative actions from Mondo conserved regions and essential trans-acting factor 14-3-3. Molecular Endocrinology. 2008 Jul 1;22(7):1658-72.
134. Arden C, Tudhope SJ, Petrie JL, Al-Oanzi ZH, Cullen KS, Lange AJ, et al. Fructose 2, 6-bisphosphate is essential for glucose-regulated gene transcription of glucose-6-phosphatase and other ChREBP target genes in hepatocytes. Biochemical Journal. 2012 Apr 1;443(1):111-23.
135. Dentin R, Tomas-Cobos L, Foufelle F, Leopold J, Girard J, Postic C, et al. Glucose 6-phosphate, rather than xylulose 5-phosphate, is required for the activation of ChREBP in response to glucose in the liver. Journal of Hepatology. 2012 Jan 1;56(1):199-209.
136. Iizuka K, Wu W, Horikawa Y, Takeda J. Role of glucose-6-phosphate and xylulose-5-phosphate in the regulation of glucose-stimulated gene expression in the pancreatic β cell line, INS-1E. Endocrine Journal. 2013;60(4):473-82.
137. Kabashima T, Kawaguchi T, Wadzinski BE, Uyeda K. Xylulose 5-phosphate mediates glucose-induced lipogenesis by xylulose 5-phosphate-activated protein phosphatase in rat liver. Proceedings of the National Academy of Sciences. 2003 Apr 29;100(9):5107-12.
138. Li MV, Chen W, Harmancey RN, Nuotio-Antar AM, Imamura M, Saha P, et al. Glucose-6-phosphate mediates activation of the carbohydrate responsive binding protein (ChREBP). Biochemical and Biophysical Research Communications. 2010 May 7;395(3):395-400.
139. Iizuka K. The transcription factor carbohydrate-response element-binding protein (ChREBP): A possible link between metabolic disease and cancer. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2017 Feb 1;1863(2):474-85.
140. Herman MA, Peroni OD, Villoria J, Schön MR, Abumrad NA, Blüher M, et al. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature. 2012 Apr 19;484(7394):333-8.
141. Li MV, Chang B, Imamura M, Poungvarin N, Chan L. Glucose-dependent transcriptional regulation by an evolutionarily conserved glucose-sensing module. Diabetes. 2006 May 1;55(5):1179-89.
142. Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proceedings of the National Academy of Sciences. 2004 May 11;101(19):7281-6.
143. Iizuka K, Miller B, Uyeda K. Deficiency of carbohydrate-activated transcription factor ChREBP prevents obesity and improves plasma glucose control in leptin-deficient (ob/ob) mice. American Journal of Physiology-Endocrinology and Metabolism. 2006 Aug;291(2):E358-64.
144. Dentin R, Benhamed F, Hainault I, Fauveau V, Foufelle F, Dyck JR, et al. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes. 2006 Aug 1;55(8):2159-70.
145. Gauthier K, Billon C, Bissler M, Beylot M, Lobaccaro JM, Vanacker JM, et al. Thyroid hormone receptor β (TRβ) and liver X receptor (LXR) regulate carbohydrate-response element-binding protein (ChREBP) expression in a tissue-selective manner. Journal of Biological Chemistry. 2010 Sep 3;285(36):28156-63.
146. Hellemans K, Grinko I, Rombouts K, Schuppan D, Geerts A. All-trans and 9-cis retinoic acid alter rat hepatic stellate cell phenotype differentially. Gut. 1999 Jul 1;45(1):134-42.
147. Hellemans K, Verbuyst P, Quartier E, Schuit F, Rombouts K, Chandraratna RA, et al. Differential modulation of rat hepatic stellate phenotype by natural and synthetic retinoids. Hepatology. 2004 Jan;39(1):97-108.
148. Wang L, Tankersley LR, Tang M, Potter JJ, Mezey E. Regulation of α2 (I) collagen expression in stellate cells by retinoic acid and retinoid X receptors through interactions with their cofactors. Archives of biochemistry and biophysics. 2004 Aug 1;428(1):92-8.
149. Lee HC, Inoue T, Imae R, Kono N, Shirae S, Matsuda S, et al. Caenorhabditis elegans mboa-7, a member of the MBOAT family, is required for selective incorporation of polyunsaturated fatty acids into phosphatidylinositol. Molecular biology of the cell. 2008 Mar;19(3):1174-84.
150. Tanaka Y, Shimanaka Y, Caddeo A, Kubo T, Mao Y, Kubota T, et al. LPIAT1/MBOAT7 depletion increases triglyceride synthesis fueled by high phosphatidylinositol turnover. Gut. 2021 Jan 1;70(1):180-93.
151. Thangapandi VR, Knittelfelder O, Brosch M, Patsenker E, Vvedenskaya O, Buch S, et al. Loss of hepatic Mboat7 leads to liver fibrosis. Gut. 2021 May 1;70(5):940-50.
152. Buch S, Stickel F, Trepo E, Way M, Herrmann A, Nischalke HD, et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nature Genetics. 2015 Dec;47(12):1443-8.
153. Mancina RM, Dongiovanni P, Petta S, Pingitore P, Meroni M, Rametta R, et al. The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology. 2016 May 1;150(5):1219-30.
154. Donati B, Dongiovanni P, Romeo S, Meroni M, McCain M, Miele L, et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Scientific Reports. 2017 Jul 3;7(1):4492.
155. Anstee QM, Darlay R, Cockell S, Meroni M, Govaere O, Tiniakos D, et al. Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically characterised cohort☆. Journal of Hepatology. 2020 Sep 1;73(3):505-15.
156. Sookoian S, Flichman D, Garaycoechea ME, Gazzi C, Martino JS, Castaño GO, Pirola CJ. Lack of evidence supporting a role of TMC4-rs641738 missense variant-MBOAT7- intergenic downstream variant-in the Susceptibility to Nonalcoholic Fatty Liver Disease. Sci Rep. 2018 Mar 23;8(1):5097.
157. Smagris E, Gilyard S, BasuRay S, Cohen JC, Hobbs HH. Inactivation of Tm6sf2, a gene defective in fatty liver disease, impairs lipidation but not secretion of very low density lipoproteins. Journal of Biological Chemistry. 2016 May 13;291(20):10659-76.
158. Li XY, Liu Z, Li L, Wang HJ, Wang H. TM6SF2 rs58542926 is related to hepatic steatosis, fibrosis and serum lipids both in adults and children: a meta-analysis. Frontiers In Endocrinology. 2022 Oct 24;13:1026901.
159. Dongiovanni P, Petta S, Maglio C, Fracanzani AL, Pipitone R, Mozzi E, et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology. 2015 Feb;61(2):506-14.
160. Pirola CJ, Sookoian S. The dual and opposite role of the TM6SF2-rs58542926 variant in protecting against cardiovascular disease and conferring risk for nonalcoholic fatty liver: A meta-analysis. Hepatology. 2015;62(6):1742-56.
161. Slosberg ED, Desai UJ, Fanelli B, St. Denny I, Connelly S, Kaleko M, et al. Treatment of type 2 diabetes by adenoviral-mediated overexpression of the glucokinase regulatory protein. Diabetes. 2001 Aug 1;50(8):1813-20.
162. Santoro N, Zhang CK, Zhao H, Pakstis AJ, Kim G, Kursawe R, et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology. 2012 Mar;55(3):781-9.
163. Raimondo A, Rees MG, Gloyn AL. Glucokinase regulatory protein: complexity at the crossroads of triglyceride and glucose metabolism. Current Opinion in Lipidology. 2015 Apr 1;26(2):88-95.
164. Petta S, Miele L, Bugianesi E, Cammà C, Rosso C, Boccia S, et al. Glucokinase regulatory protein gene polymorphism affects liver fibrosis in non-alcoholic fatty liver disease. PLoS One. 2014 Feb 3;9(2):e87523.
165. EK S. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet.. 2011;7:e1001324.
166. Zain SM, Mohamed Z, Mohamed R. A common variant in the glucokinase regulatory gene rs780094 and risk of nonalcoholic fatty liver disease: A meta‐analysis. Journal of Gastroenterology and Hepatology. 2015 Jan;30(1):21-7.
167. Perbal B. CCN proteins: multifunctional signalling regulators. The Lancet. 2004 Jan 3;363(9402):62-4.
168. Yeger H, Perbal B. The CCN family of genes: a perspective on CCN biology and therapeutic potential. Journal of Cell Communication and Signaling. 2007 Dec;1:159-64.
169. Holbourn KP, Acharya KR, Perbal B. The CCN family of proteins: structure–function relationships. Trends in Biochemical Sciences. 2008 Oct 1;33(10):461-73.
170. Zhang Y, Wang C. Nephroblastoma overexpressed (NOV/CCN3) gene: a paired-domain-specific PAX3-FKHR transcription target that promotes survival and motility in alveolar rhabdomyosarcoma cells. Oncogene. 2011 Aug;30(32):3549-62.
171. Escoté X, Gómez-Zorita S, López-Yoldi M, Milton-Laskibar I, Fernández-Quintela A, Martínez JA, et al. Role of omentin, vaspin, cardiotrophin-1, TWEAK and NOV/CCN3 in obesity and diabetes development. International Journal of Molecular Sciences. 2017 Aug 15;18(8):1770.
172. Li JY, Wang YD, Qi XY, Ran L, Hong T, Yang J, et al. Serum CCN3 levels are increased in type 2 diabetes mellitus and associated with obesity, insulin resistance and inflammation. Clin Chim Acta. 2019 Jul;494:52-57.
173. Wu W, Hu X, Zhou X, Klenotic PA, Zhou Q, Lin Z. Myeloid deficiency of CCN3 exacerbates liver injury in a mouse model of nonalcoholic fatty liver disease. J Cell Commun Signal. 2018 Mar;12(1):389-99.
174. Bugianesi E, Pagotto U, Manini R, Vanni E, Gastaldelli A, de Iasio R, et al. Plasma adiponectin in nonalcoholic fatty liver is related to hepatic insulin resistance and hepatic fat content, not to liver disease severity. The Journal of Clinical Endocrinology & Metabolism. 2005 Jun 1;90(6):3498-504.
175. Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. The Journal of Clinical Investigation. 2003 Jul 1;112(1):91-100.
176. Faggioni R, Jones-Carson J, Reed DA, Dinarello CA, Feingold KR, Grunfeld C, et al. Leptin-deficient (ob/ob) mice are protected from T cell-mediated hepatotoxicity: role of tumor necrosis factor α and IL-18. Proceedings of the National Academy of Sciences. 2000 Feb 29;97(5):2367-72.
177. Fantuzzi G, Faggioni R. Leptin in the regulation of immunity, inflammation, and hematopoiesis. Journal of Leukocyte Biology. 2000 Oct;68(4):437-46.
178. Marra F. Leptin and liver fibrosis: a matter of fat. Gastroenterology. 2002 May 1;122(5):1529-32.
179. Lehrke M, Reilly MP, Millington SC, Iqbal N, Rader DJ, Lazar MA. An inflammatory cascade leading to hyperresistinemia in humans. PLoS Medicine. 2004 Nov;1(2):e45.
180. Pagano C, Soardo G, Pilon C, Milocco C, Basan L, Milan G, et al. Increased serum resistin in nonalcoholic fatty liver disease is related to liver disease severity and not to insulin resistance. J Clin Endocrinol Metab. 2006 Mar;91(3):1081-6.
181. Ailhaud G. Adipose tissue as a secretory organ: from adipogenesis to the metabolic syndrome. Comptes Rendus Biologies. 2006 Aug 1;329(8):570-7.
182. Fantuzzi G. Adipose tissue, adipokines, and inflammation. Journal of Allergy and Clinical Immunology. 2005 May 1;115(5):911-9.
183. Schäffler A, Schölmerich J, Büchler C. Mechanisms of disease: adipocytokines and visceral adipose tissue—emerging role in nonalcoholic fatty liver disease. Nature Clinical Practice Gastroenterology & Hepatology. 2005 Jun 1;2(6):273-80.
184. Xu L, Liu W, Bai F, Xu Y, Liang X, Ma C, et al. Hepatic macrophage as a key player in fatty liver disease. Frontiers in Immunology. 2021 Dec 9;12:708978.
185. Chen W, Zhang J, Fan HN, Zhu JS. Function and therapeutic advances of chemokine and its receptor in nonalcoholic fatty liver disease. Therapeutic Advances in Gastroenterology. 2018 Dec;11:1756284818815184.
186. Lu H, Wu C, Howatt DA, Balakrishnan A, Moorleghen JJ, Chen X, et al. Angiotensinogen Exerts Effects Independent of Angiotensin II. Arterioscler Thromb Vasc Biol. 2016 Feb;36(2):256-65.
187. Ansari RA, Rizvi SAA, Husain K, Osna N. Role of Angiotensin Peptides Precursor in Ethanol Mediated Hepato toxicity: Perspective on Angiotensinogen. The Open Gastroenterology Journal. 2012;6:16-26.
188. Mohana VU, Swapna N, Surender RS, Vishnupriya S, Padma T. Gender-related association of AGT gene variants (M235T and T174M) with essential hypertension—a case-control study. Clinical and Experimental Hypertension. 2012 Feb 1;34(1):38-44.
189. Jeunemaitre X, Inoue I, Williams C, Charru A, Tichet J, Powers M, Sharma AM, Gimenez-Roqueplo AP, Hata A, Corvol P, Lalouel JM. Haplotypes of angiotensinogen in essential hypertension. Am J Hum Genet. 1997 Jun;60(6):1448-60.
190. Li Y, Jain S, Patil S, Kumar A. A haplotype of angiotensinogen gene that is associated with essential hypertension increases its promoter activity in adipocytes. Vascular Pharmacology. 2006 Jan 1;44(1):29-33.
191. Jain S, Vinukonda G, Fiering SN, Kumar A. A haplotype of human angiotensinogen gene containing− 217A increases blood pressure in transgenic mice compared with− 217G. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2008 Dec;295(6):R1849-57.
192. Padma G, Charita B, Swapna N, Mamata M, Padma T. Novel variants detected in AGT gene among patients with essential hypertension. Journal of the Renin-Angiotensin-Aldosterone System. 2015 Sep;16(3):642-6.
193. Jain S, Tillinger A, Mopidevi B, Pandey VG, Chauhan CK, Fiering SN, et al. Transgenic mice with− 6A haplotype of the human angiotensinogen gene have increased blood pressure compared with− 6G haplotype. Journal of Biological Chemistry. 2010 Dec 24;285(52):41172-86.
194. Jenkins LD, Powers RW, Cooper M, Gallaher MJ, Markovic N, Ferrell R, Ness RB, Roberts JM. Preeclampsia risk and angiotensinogen polymorphisms M235T and AGT -217 in African American and Caucasian women. Reprod Sci. 2008 Sep;15(7):696-701.
195. Sherman CT, Brasier AR. Role of signal transducers and activators of transcription 1 and-3 in inducible regulation of the human angiotensinogen gene by interleukin-6. Molecular Endocrinology. 2001 Mar 1;15(3):441-57.
196. Harrison SA, Kadakia S, Lang KA, Schenker S. Nonalcoholic steatohepatitis: what we know in the new millennium. Official Journal of the American College of Gastroenterology| ACG. 2002 Nov 1;97.
197. Saghir S, Abbas A, Alfuraih S, Sharma A, Latimer J, Omidi Y, et al. Non-alcoholic fatty liver disease: Role of PNPLA3 and its association with environmental chemicals. Arch Clin Toxicol. 2024;6(1):21-32.