Abstract
Activation of the adenylyl cyclase – PKA – CREB signaling pathway has been shown to be critical for learning and the creation and consolidation of memory. The PDE4, or cAMP-specific, phosphodiesterases (PDEs) are important modulators of cAMP levels in the central nervous system. The establishment of the essential role of PDE4 isoforms in learning and memory has been based on a long-standing interplay between experiments in genetically-tractable organisms, such as flies and mice, and pharmacological studies in rodents, primates and humans. Collectively, these diverse approaches have demonstrated the pivotal role of PDE4s in learning and memory and that pharmacological targeting of PDE4s can enhance learning, memory and cognition in humans. However, several challenges remain before PDE4 inhibitors can be used clinically in disorders of cognition and memory. Current priorities in PDE4 drug discovery focused on the CNS include 1) further refinement of isoform selectivity; 2) more precise action at CNS targets implicated in cognition and memory, such as those in forebrain and hippocampus, while reducing undesired action at other CNS (area postrema) and non-CNS targets (CFTR, gastric acid secretion) that account for the limiting side effects of many, but not all, PDE4 inhibitors in clinical trials; and 3) improved pharmacokinetics and dosing.
Keywords
Roflumilast, Apremilast, Zatolmilast, Nerandomilast, Orismilast
Genetic and Pharmacologic Evidence for PDE4 Action in Learning and Memory
Two distinct, but intertwining, threads of inquiry have demonstrated that the PDE4, or cAMP-selective, phosphodiesterases, have an essential role in human cognition, learning and memory. Study of genetically-modified preclinical models, and of humans with mutations in the PDE4D gene, has provided some of the most rigorous proof of the importance of PDE4 signaling in the CNS. More recently, clinical trials of PDE4-selective inhibitors have shown promising clinical activity in disorders of cognition and memory.
Genetic Models of PDE4 Action in the CNS
Studies in Drosophila melanogaster and aplysia
Extensive genetic study of the fruit fly Drosophila melanogaster has demonstrated a vital role for many elements of cAMP signaling in learning and short-term memory [1]. Mutations in several Drosophila genes, including rutabaga, which encodes adenylyl cyclase [2], DCO, encoding the cAMP-dependent protein kinase (protein kinase A; PKA) catalytic subunit [3], and dunce, which encodes a PDE closely homologous to mammalian PDE4 enzymes [4-8], produce clear alterations in numerous experimental tests of learning [9]. Extensive study of plasticity in the gill and siphon reflex, which serves as a measure of learning, in the marine mollusk Aplysia has demonstrated the critical role of PKA and other cAMP signaling elements in this process [10,11]; see refs. [12,13] for a review.
cAMP signaling components in learning and memory in genetically-modified mice
Several different genetic approaches have been utilized to study the roles of cAMP signaling elements in learning and memory in mice. Heterozygous and homozygous gene knockout studies have shown that knockout of specific adenylyl cyclase isoforms produces profound alterations in long-term potentiation, a key attribute of learning [14,15]. Knockouts of PKA regulatory or catalytic subunits produce defects in hippocampal long-term depression and depotentiation [16-18]. Knockouts of the cyclic nucleotide response element binding protein (CREB) also produce deficiencies in long-term memory [19,20].
As an alternative approach, expression of an inhibitory form of the PKA regulatory subunit, expressed as a transgene off the Ca++/calmodulin protein kinase IIα (CamKIIα) promoter [21], produces defects in long-term potentiation in the CA1 area of the hippocampus [22]. Similarly, expression of a dominant-negative mutant of CamKIIα, expressed off its own promoter, produces deficiencies in long-term potentiation and spatial learning [21,23-25]. Expression of a transgenic CREB with a mutation at its PKA-phosphorylation site (S133A), which serves as a dominant-negative, produces deficiencies in long-term conditioned fear memories [26,27]; see ref. [28] for a review. Collectively, these genetic studies provide compelling evidence for the role of cAMP signaling pathways in synaptic plasticity, learning and memory [29,30]. They also demonstrate that a variety of genetic approaches can be employed to study cAMP signaling in mice, with compatible results; specifically, both knockout (total animal or tissue-specific) approaches and, as an alternative, transgenic expression of a dominant-negative mutant, can successfully produce reproducible phenotypes in mice that provide insights into the mechanisms of learning, memory and cognition.
Phenotypes of mice with knockouts of PDE4D
The successful use of knockout (KO) approaches to study several different elements of cAMP signaling pathways in mice, as described above, provided an impetus to the study of the CNS phenotypes of mice with knockouts in various members of the PDE4 family. The PDE4s are members of a large superfamily of cyclic nucleotide phosphodiesterases, designed PDE1 through PDE11, which are encoded by 21 different genes. The PDE4s are encoded by 4 different genes in mammals, designated PDE4A through PDE4D in humans, with additional isoform diversity produced by alternative mRNA splicing and the use of multiple promoters in each gene; see ref. [31] for a review. Mice with knockouts of each Pde4 gene are available, with Pde4a, Pde4b and Pde4d KO mice having been studied most extensively. Pde4a-/- (homozygous KO) mice display anxiogenic-like behavior [32], but the full range of CNS phenotypes of these mice has yet to be determined. We have discussed the CNS phenotypes of Pde4b-/- mice in detail elsewhere [33]. The available data on the CNS phenotypes of Pde4d -/- mice are conflicting [34-36]. Pde4d-/- mice may have decreased immobility in forced-swim and tail-suspension tests, mimicking an antidepressant effect [35]. The Pde4d-/- genotype may improve performance in tests of learning and memory [36], although this effect was not detected in studies performed by a second group [34]. Significant non-CNS phenotypes, such as slow growth, small adult size, and impaired fertility are also seen in some Pde4d-/- mice [37,38]. More generally, the behavioral phenotypes that are typically seen in knockout mice may be highly dependent on strain background, which does not appear to have been standardized among these studies. In addition, each study employed a different subset of behavioral assays to characterize the phenotypes seen in these mice, which greatly hampers critical, side-by-side, comparison of these studies. Experimental factors, such as differences in assay conditions, sample size, or age at the time of study, could also explain some of the differing results noted to date.
The human PDE4D and mouse Pde4d genes each encode at least 11 different protein isoforms [39-43]; see ref. [44] for a review. The different isoforms differ in length, enzymatic properties, sub-cellular localization, their ability to interact with other cellular proteins, and their distribution in cells and tissues. All enzymatically-active Pde4d isoforms share an identical catalytic domain, located in the carboxyl-terminal portion of the protein, and therefore can be inhibited by PDE4-selective inhibitors. Among the best-studied PDE4D isoforms is PDE4D5, which is highly conserved among mammals and contains a unique amino-terminal domain of 88 amino acids. PDE4D5 interacts selectively with the signaling proteins RACK1 [45-47] and β-arrestin2 [48-51], and this selective interaction is mediated, at least in part, by its unique amino-terminal domain. To date, Pde4d-/- mice have been generated by targeting exons that encode the PDE4D catalytic domain, thereby disrupting all of the enzymatically-active isoforms encoded by the Pde4d gene. Therefore, Pde4d-/- mice have a phenotype that reflects the combined loss of all 11 PDE4D isoforms, which prevents study of the effect(s) of any individual isoform, such as PDE4D5. Given the presence of these multiple PDE4D isoforms, each with unique properties, it would be desirable to utilize an experimental approach that would target a single PDE4D isoform; such an approach is described in the next section.
Phenotypes of mice that express dominant-negative forms of PDE4D
We and other groups have used a dominant-negative approach to the study of the role(s) of select PDE4 isoforms in the CNS. This approach is conceptually identical to the use of dominant-negative or inhibitory mutants of PKA, CamKIIα, or CREB as genetic probes of cAMP signaling in the CNS, as described in the studies cited above. The PDE4 studies have used engineered versions of select PDE4 isoforms with mutations of an aspartate deep in the catalytic domain that is responsible for metal-binding and therefore catalysis [48,49]. Mutation of this aspartate totally abolishes the catalytic activity of the isoform, with no detectable effect on the stability of the protein, its ability to be targeted to specific sub-cellular locations, or its interactions with other proteins. Instead, the mutant serves to displace the corresponding endogenous PDE4 isoform from its protein partner(s), thereby preventing the hydrolysis of cAMP in the specific sub-cellular compartment(s) where that isoform is normally expressed.
Havekes, Vecsey and their colleagues have demonstrated that expression of the PDE4A5-D577A mutant (also called PDE4D5-catnull), expressed in the mouse hippocampus off the CamKIIα promoter, using a viral construct, reverses or attenuates several protein-phosphorylation events that are associated with sleep deprivation [52-54]. It also prevents or reduces learning/memory deficits that are normally associated with sleep deprivation [54]. No such effect was seen with expression of a double mutant, PDE4A5-catnullΔ4, containing both the D577A mutation and removal of the unique PDE4A5 amino-terminal domain, which is responsible for its intracellular targeting and/or interactions with other signaling components. These data demonstrate the value of a dominant-negative experimental approach to the study of the functional roles of specific PDE4 isoforms in the CNS, with particular emphasis on learning and memory.
Previously, we have utilized a similar approach to characterize the functions of a PDE4B isoform, specifically PDE4B1, one of 5 isoforms, all highly conserved, encoded by the human PDE4B and mouse Pde4b genes [33]. The PDE4B1-D564A mutation, at a highly-conserved catalytic domain aspartate homologous to PDE4A5-D577 (see preceding paragraph), was expressed as a transgene under the control of the CamKIIα promoter. The expression of genes under the control of this promoter is preferentially limited to adult forebrain excitatory neurons, including those in the hippocampus, amygdala, cortex, and striatum [21]. Mice expressing the PDE4B1-D564A transgene showed increased phosphorylation of CREB and ERK1/2, increased hippocampal neurogenesis, and enhanced baseline synaptic transmission and long-term potentiation (LTP; ref. [33]). Our data supported the use of a dominant-negative, isoform-selective approach to the study of PDE4B1 function in the CNS and the role of PDE4B isoforms in CNS functions.
These studies with PDE4A5-D577A and PDE4B1-D564A provided an impetus to the use of a conceptually-similar approach to PDE4D isoforms, most notably PDE4D5; ref. [44]. We therefore generated transgenic mice with the critical metal-binding aspartate mutation, PDE4D5-D556A, expressed off the CamKIIα promoter [44]. We chose PDE4D5 because (1) human PDE4D5 (GenBank AF012073) and mouse PDE4D5 (GenBank XP_006517707.1) are extremely similar, being identical in length and having a 98% amino acid identity [39]; (2) PDE4D5 mRNA is expressed in several regions of the mouse CNS [55, 56] compatible with it being targeted by a transgene driven by the CamKIIα promoter; (3) PDE4D5 interacts with several important signaling proteins, most notably RACK1 and β-arrestin2, as outlined above. PDE4D5 appears to have a pivotal role in regulating signaling through the β2-adrenergic receptor [48,49,57-59] and therefore these experiments might provide insights into its role(s) in β2-adrenergic signaling in the CNS.
When overexpressed in cell-based systems, PDE4D5-D556A produces no change in total PDE4 enzymatic activity, but instead disrupts PDE4D5 cellular function in a dominant-negative fashion by producing an equilibrium displacement of endogenous PDE4D5 from its protein partner(s), most notably RACK1 and β-arrestin2 [48,49,51]. In our mouse model, the PDE4D5-D556A transgene appears to have acted in a dominant-negative fashion, blocking PDE4D5 function in regions of the CNS where PDE4D5 is active, and increasing cAMP levels. Elevation of cAMP activated PKA, leading to enhanced phosphorylation of CREB, and/or stimulated other cAMP effectors, such as EPAC [60-62] or cyclic nucleotide-gated ion channels [63]. These putative changes in cAMP signaling were associated with significant alterations in behavior, most notably affecting hippocampal-dependent positional learning (place preference), based on their performance in the Morris water maze [44]. In contrast, the PDE4D5-D556A transgene had no detectable effect on associative fear conditioning. A marked, sex-dependent effect of the transgene on behavior in an open field test was also observed [44].
The extensive experimentation that has been performed with the dominant-negative mouse models has a number of methodological limitations. First of all, for the transgene to produce the desired effect in the CNS, it needs to be expressed at a level sufficiently high to displace all, or at least a functionally significant proportion, of the wild-type PDE protein from its binding partners. In the reported studies, expression of the transgene in brain tissues was typically performed using immunoblotting; cell-based functional assays were not felt to be feasible in brain tissue. Therefore, some uncertainty remains whether these mutants were indeed acting in a true dominant-negative fashion in the brain. There may also be significant methodological differences in the assays employed by the two groups performing these studies. Other issues, such as sample size, treatment of the mice prior to the performance of the assays, or age at the time of study, may also complicate critical analysis of their results.
Promising Human Clinical Trials with Roflumilast
Following the pioneering work of Barad [64] and Bach [65] on the pro-cognitive effects of PDE4 inhibitors in pre-clinical models, a series of human clinical trials has demonstrated the value of roflumilast as an enhancer of cognitive function in both healthy adults and those with age-related memory dysfunction [66-71]. Roflumilast is a PDE4 inhibitor currently marketed in oral form for chronic obstructive pulmonary disease (COPD) and as a topical formulation for inflammatory skin disorders, but has yet to receive marketing approval for any CNS indication. Clinical use of roflumilast in CNS indications is likely to be limited by typical side effects of the drug, including nausea, emesis and diarrhea [72,73]. These side effects are typical of most 1st- and 2nd-generation PDE4 selective inhibitors (i.e., are class-specific). Nausea/emesis has probably been the most commonly-encountered limiting side effect in clinical trials of these agents, and reflects action at both CNS (i.e., area postrema) and non-CNS (gastric acid secretion, GI motility) targets. The non-infectious diarrhea that is seen in many patients with roflumilast probably reflects its activation of CFTR, a Cl- ion channel, in GI mucosa [72]. The generation of PDE4 inhibitors lacking these side effects is a major emphasis in PDE4 drug discovery, as will be described in the next sections.
Implications for CNS Drug Discovery – Potential Obstacles
Precision targeting of the PDE4 catalytic region
Clinically-available PDE4 inhibitors, specifically roflumilast, apremilast and crisaborole, act at the catalytic domain of the PDE4 enzymes, where they serve, at least in part, as competitive inhibitors of cAMP hydrolysis [74-76]. Given the high degree of homology among the catalytic domains of the enzymes encoded by the 4 PDE4 genes, which have at least 90% amino acid sequence homology [7,31,77,78], it is not surprising that these 3 drugs, as well as the vast majority of PDE4-selective inhibitors that have been synthesized to date, have similar potencies against isoforms encoded by all 4 PDE4 genes (i.e., IC50 values differing by less than 10-fold). For example, GEBR-7b and GEBR-32a, which has been extensively tested against several different PDE4D isoforms, has a difference in IC50 for PDE4D isoforms, as compared to PDE4A or PDE4B isoforms, of less than 10-fold [79-82]. These data suggest that exclusive targeting of the PDE4 catalytic domains is unlikely to produce the specificity of action required for selectivity for different isoforms encoded by any one PDE4 gene.
Importance of isoform specificity
Among the important lessons gained from studies employing dominant-negative transgenes is the importance of isoform specificity. Although each of the PDE4A, PDE4B, and PDE4D genes encodes multiple isoforms, the 3 studies summarized above each focused on the effects of a single isoform encoded by each of these genes. This experimental design contrasts with gene knockout studies, which affect all the isoforms encoded by each gene. Similarly, the studies using gene “knockdowns” with lentiviral siRNAs as probes of PDE4 function in the CNS have used constructs that appear to target multiple isoforms [36,83-89]. The significant phenotypes produced by targeting individual isoforms in the dominant-negative studies provides strong functional evidence that each isoform has an important and unique biological function. It is unlikely that the full therapeutic potential of PDE4 inhibition will be reached until agents are developed that can target individual isoforms with a high degree of selectivity.
Importance of interactions with PDE4 partner proteins
Among the best-studied properties of PDE4 proteins is their interactions with a wide range of other cellular proteins. For example, PDE4A5 interacts selectively with the immunophilin homolog AIP1 (also known as XAP2, ref. [90]). PDE4B1, and probably other PDE4B isoforms, may functionally interact with the DISC1 protein [91-101]. As mentioned above, PDE4D5 interacts with RACK1 and β-arrestin2, as well as potentially other proteins. These protein-protein interactions have the potential to influence the folding of select PDE4 isoforms, their enzymatic properties, sensitivity to PDE4 inhibitors, their sub-cellular localization, and their ability to interact with other proteins. It is highly likely that many of the effects of the dominant-negative mutants, when expressed in the CNS, reflect alterations in their interactions with their associated partner proteins. For example, by dramatically increasing scaffold sites for β-arrestin2 and RACK1, PDE4D5-D556A may sequester these proteins and thereby block their normal functions in cells. This effect would produce a CNS phenotype that would differ from that seen with small-molecule inhibitors of the catalytic activity of PDE4D5. Finally, “long” PDE4 isoforms can form stable dimers that are regulated, at least in part, by PKA phosphorylation [102]; these important protein-protein interactions clearly can be affected by expression of our dominant-negative mutants, but has yet to be targeted convincingly by small-molecule agents.
Implications for PDE4 Drug Discovery – Potential Solutions
The potential value of allosteric modulators
Among the most important recent developments in PDE4 pharmacology is the development of compounds that allosterically target PDE4 isoforms (Figure 1). These compounds target PDE4 isoforms by interacting, at least in part, with their amino-terminal regulatory domains, UCR1 and UCR2; ref. [7]. Long PDE4 isoforms contain UCR1, UCR2 and the catalytic domain, while short isoforms lack UCR1, and super-short isoforms lack UCR1 and the amino-terminal half of UCR2. The dimerization of long PDE4 isoforms requires a trans-activation between UCR1 and UCR2 [102-106]. This process is regulated, at least in part, by PKA, which phosphorylates these isoforms at a conserved serine located at the amino-terminal end of UCR1 [107,108] and increases their catalytic activity (relative Vmax). Intriguingly, mutations in amino acids in PDE4D UCR1 or UCR2 that are critical for PKA action and dimerization have been shown to cause acrodysostosis, an inherited human disorder of bone that is also associated with significant intellectual and cognitive dysfunction [102,109,110].
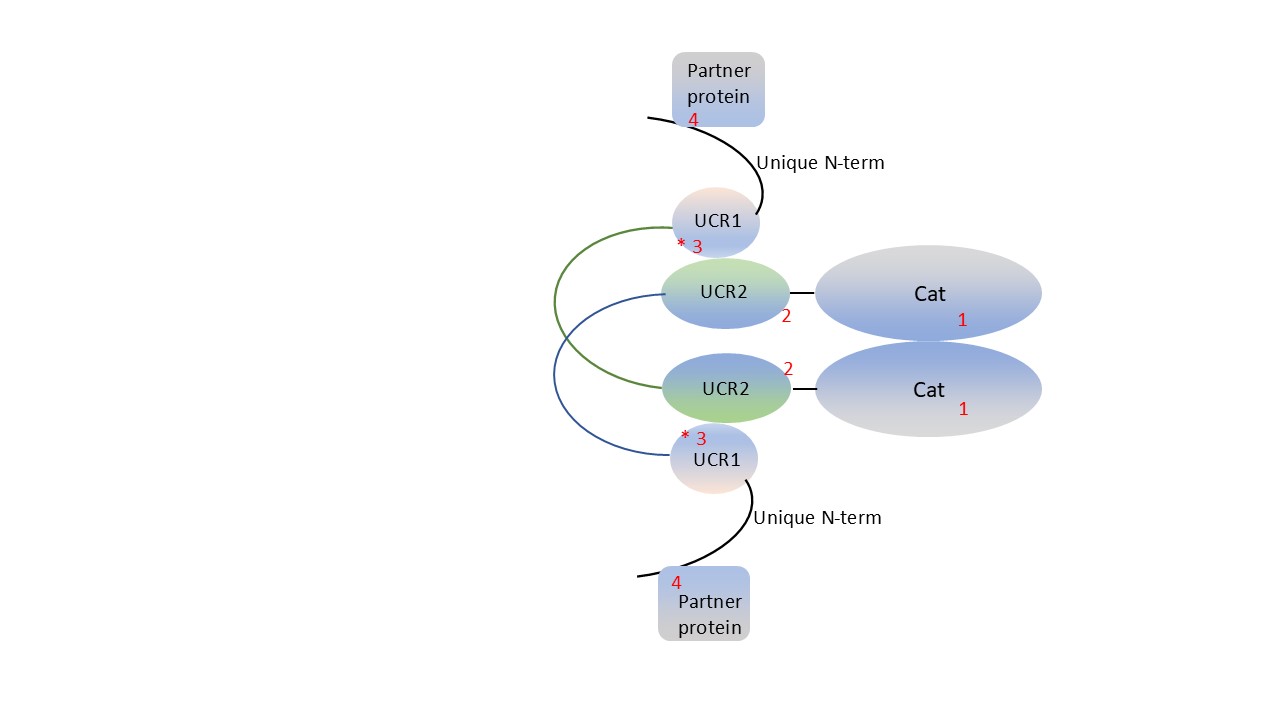
Figure 1:Schematic of PDE4 structure and potential sites for drug action. Long PDE4 isoforms, as shown here, contain UCR1, UCR2 and the catalytic domains of the enzyme. These 3 domains are linked by short, unstructured regions (shown as thin lines). Dimerization of long PDE4 isoforms is driven by trans-interaction between UCR1 and UCR2 and also by direct interaction between the catalytic domains. Classic competitive PDE4 inhibitors act on the catalytic domain (1) by displacing its physiologic substrate, cAMP. PDE4 allosteric modulators commonly act on a region of UCR2 that is located close to the catalytic domain (2) and, in many cases, act directly on the catalytic domain (1). Additional potential sites for drug action include regions of UCR1 and/or UCR2 that are required for dimerization (3), which may also affect the ability of the enzyme to be regulated by phosphorylation by PKA (asterisk, *) and potentially by other kinases. Finally, specific PDE4 isoforms bind, and can be regulated by, interactions with other proteins (e.g., RACK1 or β-arrestin2, both of which interact, at least in part, with the unique amino-terminal region of PDE4D5); these interactions may provide additional potential sites for drug action (4).
The interactions between UCR1, UCR2 and the catalytic region of long PDE4 isoforms provides the mechanistic basis for the development of allosteric modulators of PDE4 enzymes. These compounds typically interact with both the catalytic domain and the UCR1/2 domains, but, in the case of PDE4 allosteric activators, may interact solely with UCR1/2 [111-117]; see ref. [118] for a review. Given the large size of these molecules, and the resulting complex interactions with larger region(s) of the PDE4 dimer, it is perhaps not surprising that they show significant selectivity as inhibitors. For example, DI159687 (and several closely related compounds) has approximately a 20- to 54-fold selectivity (IC50) for human long PDE4D isoforms, as opposed to human long PDE4B isoforms [111]. Similarly, zatolmilast (previously known as BPN14770) has approximately 200-fold selectivity for dimeric PDE4D, compared to dimeric PDE4B [115]. Zatolmilast is currently being tested in multiple human clinical trials for Fragile X Syndrome. NCS 613 appears to have selectivity for PDE4C [119-121]. Conversely, A-33 is approximately 100-fold selective for PDE4B isoforms, compared to PDE4D isoforms [122], although the relative IC50 data on this compound has been reported only by a single group (It has yet to be shown that A-33 interacts with UCR1/UCR2, in addition to the catalytic domain, consistent with a true allosteric modulator, although its structure is compatible with such an interaction [115].). The compound FCPR16, and the related compound 4e, also have significant selectivity for PDE4B [123-125]. Orismilast also has modest, although possibly significant, selectivity for PDE4B [126]. KVA-D-88 is also about 6-fold selective for PDE4B [127]. Nerandomilast (previously known as BI101550) shows selectivity for PDE4B that is 9-fold, 24.8-fold, and 870-fold greater than for PDE4D, PDE4A, and PDE4C, respectively [128]. The IC50 data for nerandomilast were reported only for short PDE4 isoforms; i.e., lacking UCR1, so it is uncertain whether this compound is a competitive inhibitor or an allosteric modulator. Nerandomilast was recently reported to meet key endpoints in a Phase III human clinical trial for idiopathic pulmonary fibrosis (IPF). The allosteric activator described by Mironid selectively activates long PDE4 isoforms via its interaction with UCR1/2 [117]. Collectively, these drug discovery efforts provide evidence that targeting of UCR1/2, in addition, or instead of, the catalytic domain, has the potential to target PDE4 isoforms selectively, using any of the following criteria: long forms v short forms, dimeric v monomeric forms; PKA-activated v basal forms; or long isoforms encoded by a specific PDE4 gene v all long PDE4 isoforms. In addition, targeting UCR1/2 can mimic critical PDE4 regulatory events, such as PKA phosphorylation [117].
The impressive number of new compounds with activity against various PDE4 isoforms is testimony to the current high level of activity in this field, but it creates significant short-term difficulties in attempting any comparative analysis of these very different chemical entities. In the majority of cases, the pre-clinical aspects of each of these compounds has been studied only by a single, industry-led, group. The types of enzymatic and functional assays, and even the exact end-points of these assays, varies substantially among these groups. Much of this reflects differences in the intended clinical uses of these modulators, which varies substantially, as outlined above. Many of these compounds have yet to enter clinical trials for any indication, and the majority of the clinical trials that have been performed to date have been focused on non-CNS indications. This situation is likely to improve with time: further studies of these compounds in pre-clinical CNS models, and then in appropriately-designed clinical trials in disorders of memory and cognition, will clarify the value of each of these compounds in the future.
Conclusions
Current momentum in PDE4 research has been propelled by a dynamic interplay between the study of genetically-modified pre-clinical models and the development of pharmacologic agents that target specific PDE4 isoforms with novel mechanisms of action. There are obvious parallels between modifying the action of PDE4s by genetic means and by many of these novel agents. Future developments are highly likely to enhance the value of both genetic tools and drug discovery. They will also continue to validate PDE4 as an experimental and therapeutic focus. Given their diverse pharmacologic effects, and the results already obtained from the genetic models, it is entirely possible that some of the newer PDE4 compounds will have activity in a variety of human CNS disorders, including those of cognition, learning and memory.
References
2. Bellen HJ, Gregory BK, Olsson CL, Kiger JA Jr. Two Drosophila learning mutants, dunce and rutabaga, provide evidence of a maternal role for cAMP on embryogenesis. Dev Biol. 1987 Jun;121(2):432-44.
3. Skoulakis EM, Kalderon D, Davis RL. Preferential expression in mushroom bodies of the catalytic subunit of protein kinase A and its role in learning and memory. Neuron. 1993 Aug;11(2):197-208.
4. Chen CN, Denome S, Davis RL. Molecular analysis of cDNA clones and the corresponding genomic coding sequences of the Drosophila dunce+ gene, the structural gene for cAMP phosphodiesterase. Proc Natl Acad Sci U S A. 1986 Dec;83(24):9313-7.
5. Davis RL, Takayasu H, Eberwine M, Myres J. Cloning and characterization of mammalian homologs of the Drosophila dunce+ gene. Proc Natl Acad Sci U S A. 1989 May;86(10):3604-8.
6. Henkel-Tigges J, Davis RL. Rat homologs of the Drosophila dunce gene code for cyclic AMP phosphodiesterases sensitive to rolipram and RO 20-1724. Mol Pharmacol. 1990 Jan;37(1):7-10.
7. Bolger G, Michaeli T, Martins T, St John T, Steiner B, Rodgers L, et al. A family of human phosphodiesterases homologous to the dunce learning and memory gene product of Drosophila melanogaster are potential targets for antidepressant drugs. Mol Cell Biol. 1993 Oct;13(10):6558-71.
8. Byers D, Davis RL, Kiger JA Jr. Defect in cyclic AMP phosphodiesterase due to the dunce mutation of learning in Drosophila melanogaster. Nature. 1981 Jan 1;289(5793):79-81.
9. Davis RL. physiology and Biochemistry of Drosophila Learning Mutants. Physiol Rev. 1996;76(2):299-317.
10. Bergold PJ, Sweatt JD, Winicov I, Weiss KR, Kandel ER, Schwartz JH. Protein synthesis during acquisition of long-term facilitation is needed for the persistent loss of regulatory subunits of the Aplysia cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1990 May;87(10):3788-91.
11. Bartsch D, Ghirardi M, Skehel PA, Karl KA, Herder SP, Chen M, et al. Aplysia CREB2 represses long-term facilitation: relief of repression converts transient facilitation into long-term functional and structural change. Cell. 1995 Dec 15;83(6):979-92.
12. Frank DA, Greenberg ME. CREB: a mediator of long-term memory from mollusks to mammals. Cell. 1994 Oct 7;79(1):5-8.
13. Kandel ER, Schwartz JH. Molecular biology of learning: modulation of transmitter release. Science. 1982 Oct 29;218(4571):433-43.
14. Wu ZL, Thomas SA, Villacres EC, Xia Z, Simmons ML, Chavkin C, et al. Altered behavior and long-term potentiation in type I adenylyl cyclase mutant mice. Proc Natl Acad Sci U S A. 1995 Jan 3;92(1):220-4.
15. Storm DR, Hansel C, Hacker B, Parent A, Linden DJ. Impaired cerebellar long-term potentiation in type I adenylyl cyclase mutant mice. Neuron. 1998 Jun;20(6):1199-210.
16. Brandon EP, Zhuo M, Huang YY, Qi M, Gerhold KA, Burton KA, et al. Hippocampal long-term depression and depotentiation are defective in mice carrying a targeted disruption of the gene encoding the RI beta subunit of cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1995 Sep 12;92(19):8851-5.
17. Huang YY, Kandel ER, Varshavsky L, Brandon EP, Qi M, Idzerda RL, et al. A genetic test of the effects of mutations in PKA on mossy fiber LTP and its relation to spatial and contextual learning. Cell. 1995 Dec 29;83(7):1211-22.
18. Qi M, Zhuo M, Skålhegg BS, Brandon EP, Kandel ER, McKnight GS, et al. Impaired hippocampal plasticity in mice lacking the Cbeta1 catalytic subunit of cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1996 Feb 20;93(4):1571-6.
19. Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell. 1994 Oct 7;79(1):59-68.
20. Kogan JH, Frankland PW, Blendy JA, Coblentz J, Marowitz Z, Schütz G, et al. Spaced training induces normal long-term memory in CREB mutant mice. Curr Biol. 1997 Jan 1;7(1):1-11.
21. Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996 Dec 6;274(5293):1678-83.
22. Abel T, Nguyen PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R. Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell. 1997 Mar 7;88(5):615-26.
23. Rotenberg A, Mayford M, Hawkins RD, Kandel ER, Muller RU. Mice expressing activated CaMKII lack low frequency LTP and do not form stable place cells in the CA1 region of the hippocampus. Cell. 1996 Dec 27;87(7):1351-61.
24. Mayford M, Wang J, Kandel ER, O'Dell TJ. CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP. Cell. 1995 Jun 16;81(6):891-904.
25. Cho YH, Giese KP, Tanila H, Silva AJ, Eichenbaum H. Abnormal hippocampal spatial representations in alphaCaMKIIT286A and CREBalphaDelta- mice. Science. 1998 Feb 6;279(5352):867-9.
26. Kida S, Josselyn SA, Peña de Ortiz S, Kogan JH, Chevere I, Masushige S, et al. CREB required for the stability of new and reactivated fear memories. Nat Neurosci. 2002 Apr;5(4):348-55.
27. Barco A, Alarcon JM, Kandel ER. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell. 2002 Mar 8;108(5):689-703.
28. Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001 Nov 2;294(5544):1030-8.
29. Kandel ER, Dudai Y, Mayford MR. The molecular and systems biology of memory. Cell. 2014 Mar 27;157(1):163-86.
30. Houslay MD, Sullivan M, Bolger GB. The multienzyme PDE4 cyclic adenosine monophosphate-specific phosphodiesterase family: intracellular targeting, regulation, and selective inhibition by compounds exerting anti-inflammatory and antidepressant actions. Adv Pharmacol. 1998;44:225-342.
31. Hansen RT 3rd, Conti M, Zhang HT. Mice deficient in phosphodiesterase-4A display anxiogenic-like behavior. Psychopharmacology (Berl). 2014 Aug;231(15):2941-54.
32. Campbell SL, van Groen T, Kadish I, Smoot LHM, Bolger GB. Altered phosphorylation, electrophysiology, and behavior on attenuation of PDE4B action in hippocampus. BMC Neurosci. 2017 Dec 2;18(1):77.
33. Rutten K, Misner DL, Works M, Blokland A, Novak TJ, Santarelli L, et al. Enhanced long-term potentiation and impaired learning in phosphodiesterase 4D-knockout (PDE4D) mice. Eur J Neurosci. 2008 Aug;28(3):625-32.
34. Zhang HT, Huang Y, Jin SL, Frith SA, Suvarna N, Conti M, et al. Antidepressant-like profile and reduced sensitivity to rolipram in mice deficient in the PDE4D phosphodiesterase enzyme. Neuropsychopharmacology. 2002 Oct;27(4):587-95.
35. Li YF, Cheng YF, Huang Y, Conti M, Wilson SP, O'Donnell JM, et al. Phosphodiesterase-4D knock-out and RNA interference-mediated knock-down enhance memory and increase hippocampal neurogenesis via increased cAMP signaling. J Neurosci. 2011 Jan 5;31(1):172-83.
36. Jin SL, Richard FJ, Kuo WP, D'Ercole AJ, Conti M. Impaired growth and fertility of cAMP-specific phosphodiesterase PDE4D-deficient mice. Proc Natl Acad Sci U S A. 1999 Oct 12;96(21):11998-2003.
37. Jin SL, Conti M. Induction of the cyclic nucleotide phosphodiesterase PDE4B is essential for LPS-activated TNF-alpha responses. Proc Natl Acad Sci U S A. 2002 May 28;99(11):7628-33.
38. Bolger GB, Erdogan S, Jones RE, Loughney K, Scotland G, Hoffmann R, et al. Characterization of five different proteins produced by alternatively spliced mRNAs from the human cAMP-specific phosphodiesterase PDE4D gene. Biochem J. 1997 Dec 1;328 ( Pt 2)(Pt 2):539-48.
39. Wang D, Deng C, Bugaj-Gaweda B, Kwan M, Gunwaldsen C, Leonard C, et al. Cloning and characterization of novel PDE4D isoforms PDE4D6 and PDE4D7. Cell Signal. 2003 Sep;15(9):883-91.
40. Gretarsdottir S, Thorleifsson G, Reynisdottir ST, Manolescu A, Jonsdottir S, Jonsdottir T, et al. The gene encoding phosphodiesterase 4D confers risk of ischemic stroke. Nat Genet. 2003 Oct;35(2):131-8.
41. Chandrasekaran A, Toh KY, Low SH, Tay SK, Brenner S, Goh DL. Identification and characterization of novel mouse PDE4D isoforms: molecular cloning, subcellular distribution and detection of isoform-specific intracellular localization signals. Cell Signal. 2008 Jan;20(1):139-53.
42. Lynex CN, Li Z, Chen ML, Toh KY, Low RW, Goh DL, et al. Identification and molecular characterization of a novel PDE4D11 cAMP-specific phosphodiesterase isoform. Cell Signal. 2008 Dec;20(12):2247-55.
43. Bolger GB, Smoot LHM, van Groen T. Dominant-Negative Attenuation of cAMP-Selective Phosphodiesterase PDE4D Action Affects Learning and Behavior. Int J Mol Sci. 2020 Aug 9;21(16):5704.
44. Yarwood SJ, Steele MR, Scotland G, Houslay MD, Bolger GB. The RACK1 signaling scaffold protein selectively interacts with the cAMP-specific phosphodiesterase PDE4D5 isoform. J Biol Chem. 1999 May 21;274(21):14909-17.
45. Bolger GB, Baillie GS, Li X, Lynch MJ, Herzyk P, Mohamed A, et al. Scanning peptide array analyses identify overlapping binding sites for the signalling scaffold proteins, beta-arrestin and RACK1, in cAMP-specific phosphodiesterase PDE4D5. Biochem J. 2006 Aug 15;398(1):23-36.
46. Bolger GB. RACK1 and β-arrestin2 attenuate dimerization of PDE4 cAMP phosphodiesterase PDE4D5. Cell Signal. 2016 Jul;28(7):706-12.
47. Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, et al. Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science. 2002 Oct 25;298(5594):834-6.
48. Baillie GS, Sood A, McPhee I, Gall I, Perry SJ, Lefkowitz RJ, et al. beta-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates beta-adrenoceptor switching from Gs to Gi. Proc Natl Acad Sci U S A. 2003 Feb 4;100(3):940-5.
49. Bolger GB, McCahill A, Huston E, Cheung YF, McSorley T, Baillie GS, et al. The unique amino-terminal region of the PDE4D5 cAMP phosphodiesterase isoform confers preferential interaction with beta-arrestins. J Biol Chem. 2003 Dec 5;278(49):49230-8.
50. Lynch MJ, Baillie GS, Mohamed A, Li X, Maisonneuve C, Klussmann E, et al. RNA silencing identifies PDE4D5 as the functionally relevant cAMP phosphodiesterase interacting with beta arrestin to control the protein kinase A/AKAP79-mediated switching of the beta2-adrenergic receptor to activation of ERK in HEK293B2 cells. J Biol Chem. 2005 Sep 30;280(39):33178-89.
51. Vecsey CG, Baillie GS, Jaganath D, Havekes R, Daniels A, Wimmer M, et al. Sleep deprivation impairs cAMP signalling in the hippocampus. Nature. 2009 Oct 22;461(7267):1122-5.
52. Havekes R, Park AJ, Tolentino RE, Bruinenberg VM, Tudor JC, Lee Y, et al. Compartmentalized PDE4A5 Signaling Impairs Hippocampal Synaptic Plasticity and Long-Term Memory. J Neurosci. 2016 Aug 24;36(34):8936-46.
53. Havekes R, Park AJ, Tudor JC, Luczak VG, Hansen RT, Ferri SL, et al. Sleep deprivation causes memory deficits by negatively impacting neuronal connectivity in hippocampal area CA1. Elife. 2016 Aug 23;5:e13424.
54. Miró X, Pérez-Torres S, Puigdomènech P, Palacios JM, Mengod G. Differential distribution of PDE4D splice variant mRNAs in rat brain suggests association with specific pathways and presynaptical localization. Synapse. 2002 Sep 15;45(4):259-69.
55. Mori F, Pérez-Torres S, De Caro R, Porzionato A, Macchi V, Beleta J, et al. The human area postrema and other nuclei related to the emetic reflex express cAMP phosphodiesterases 4B and 4D. J Chem Neuroanat. 2010 Sep;40(1):36-42.
56. Berthouze-Duquesnes M, Lucas A, Saulière A, Sin YY, Laurent AC, Galés C, et al. Specific interactions between Epac1, β-arrestin2 and PDE4D5 regulate β-adrenergic receptor subtype differential effects on cardiac hypertrophic signaling. Cell Signal. 2013 Apr;25(4):970-80.
57. Rampersad SN, Ovens JD, Huston E, Umana MB, Wilson LS, Netherton SJ, et al. Cyclic AMP phosphodiesterase 4D (PDE4D) Tethers EPAC1 in a vascular endothelial cadherin (VE-Cad)-based signaling complex and controls cAMP-mediated vascular permeability. J Biol Chem. 2010 Oct 29;285(44):33614-22.
58. Richter W, Day P, Agrawal R, Bruss MD, Granier S, Wang YL, et al. Signaling from beta1- and beta2-adrenergic receptors is defined by differential interactions with PDE4. EMBO J. 2008 Jan 23;27(2):384-93.
59. Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol. 2010;50:355-75.
60. Yang Y, Shu X, Liu D, Shang Y, Wu Y, Pei L, et al. EPAC null mutation impairs learning and social interactions via aberrant regulation of miR-124 and Zif268 translation. Neuron. 2012 Feb 23;73(4):774-88.
61. Srivastava DP, Jones KA, Woolfrey KM, Burgdorf J, Russell TA, Kalmbach A, et al. Social, communication, and cortical structural impairments in Epac2-deficient mice. J Neurosci. 2012 Aug 22;32(34):11864-78.
62. Kaupp UB, Seifert R. Cyclic nucleotide-gated ion channels. Physiol Rev. 2002 Jul;82(3):769-824.
63. Barad M, Bourtchouladze R, Winder DG, Golan H, Kandel E. Rolipram, a type IV-specific phosphodiesterase inhibitor, facilitates the establishment of long-lasting long-term potentiation and improves memory. Proc Natl Acad Sci U S A. 1998 Dec 8;95(25):15020-5.
64. Bach ME, Barad M, Son H, Zhuo M, Lu YF, Shih R, et al. Age-related defects in spatial memory are correlated with defects in the late phase of hippocampal long-term potentiation in vitro and are attenuated by drugs that enhance the cAMP signaling pathway. Proc Natl Acad Sci U S A. 1999 Apr 27;96(9):5280-5.
65. Van Duinen MA, Sambeth A, Heckman PRA, Smit S, Tsai M, Lahu G, et al. Acute administration of roflumilast enhances immediate recall of verbal word memory in healthy young adults. Neuropharmacology. 2018 Mar 15;131:31-8.
66. Heckman PRA, Van Duinen MA, Blokland A, Uz T, Prickaerts J, Sambeth A. Acute administration of roflumilast enhances sensory gating in healthy young humans in a randomized trial. Psychopharmacology (Berl). 2018 Jan;235(1):301-8.
67. Prickaerts J, Heckman PRA, Blokland A. Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer's disease. Expert Opin Investig Drugs. 2017 Sep;26(9):1033-48.
68. Heckman PR, Wouters C, Prickaerts J. Phosphodiesterase inhibitors as a target for cognition enhancement in aging and Alzheimer's disease: a translational overview. Curr Pharm Des. 2015;21(3):317-31.
69. Blokland A, Van Duinen MA, Sambeth A, Heckman PRA, Tsai M, Lahu G, et al. Acute treatment with the PDE4 inhibitor roflumilast improves verbal word memory in healthy old individuals: a double-blind placebo-controlled study. Neurobiol Aging. 2019 May;77:37-43.
70. Gilleen J, Nottage J, Yakub F, Kerins S, Valdearenas L, Uz T, et al. The effects of roflumilast, a phosphodiesterase type-4 inhibitor, on EEG biomarkers in schizophrenia: A randomised controlled trial. J Psychopharmacol. 2021 Jan;35(1):15-22.
71. Calverley PM, Rabe KF, Goehring UM, Kristiansen S, Fabbri LM, Martinez FJ; M2-124 and M2-125 study groups. Roflumilast in symptomatic chronic obstructive pulmonary disease: two randomised clinical trials. Lancet. 2009 Aug 29;374(9691):685-94.
72. Fabbri LM, Calverley PM, Izquierdo-Alonso JL, Bundschuh DS, Brose M, Martinez FJ, et al; M2-127 and M2-128 study groups. Roflumilast in moderate-to-severe chronic obstructive pulmonary disease treated with longacting bronchodilators: two randomised clinical trials. Lancet. 2009 Aug 29;374(9691):695-703.
73. Zhang KY, Card GL, Suzuki Y, Artis DR, Fong D, Gillette S, et al. A glutamine switch mechanism for nucleotide selectivity by phosphodiesterases. Mol Cell. 2004 Jul 23;15(2):279-86.
74. Huai Q, Sun Y, Wang H, Macdonald D, Aspiotis R, Robinson H, et al. Enantiomer discrimination illustrated by the high resolution crystal structures of type 4 phosphodiesterase. J Med Chem. 2006 Mar 23;49(6):1867-73.
75. Wang H, Peng MS, Chen Y, Geng J, Robinson H, Houslay MD, et al. Structures of the four subfamilies of phosphodiesterase-4 provide insight into the selectivity of their inhibitors. Biochem J. 2007 Dec 1;408(2):193-201.
76. Swinnen JV, Joseph DR, Conti M. Molecular cloning of rat homologues of the Drosophila melanogaster dunce cAMP phosphodiesterase: evidence for a family of genes. Proc Natl Acad Sci U S A. 1989 Jul;86(14):5325-9.
77. Bolger GB, Rodgers L, Riggs M. Differential CNS expression of alternative mRNA isoforms of the mammalian genes encoding cAMP-specific phosphodiesterases. Gene. 1994 Nov 18;149(2):237-44.
78. Bruno O, Romussi A, Spallarossa A, Brullo C, Schenone S, Bondavalli F, et al. New selective phosphodiesterase 4D inhibitors differently acting on long, short, and supershort isoforms. J Med Chem. 2009 Nov 12;52(21):6546-57.
79. Brullo C, Massa M, Rocca M, Rotolo C, Guariento S, Rivera D, et al. Synthesis, biological evaluation, and molecular modeling of new 3-(cyclopentyloxy)-4-methoxybenzaldehyde O-(2-(2,6-dimethylmorpholino)-2-oxoethyl) Oxime (GEBR-7b) related phosphodiesterase 4D (PDE4D) inhibitors. J Med Chem. 2014 Aug 28;57(16):7061-72.
80. Brullo C, Ricciarelli R, Prickaerts J, Arancio O, Massa M, Rotolo C, et al. New insights into selective PDE4D inhibitors: 3-(Cyclopentyloxy)-4-methoxybenzaldehyde O-(2-(2,6-dimethylmorpholino)-2-oxoethyl) oxime (GEBR-7b) structural development and promising activities to restore memory impairment. Eur J Med Chem. 2016 Nov 29;124:82-102.
81. Ricciarelli R, Brullo C, Prickaerts J, Arancio O, Villa C, Rebosio C, et al. Memory-enhancing effects of GEBR-32a, a new PDE4D inhibitor holding promise for the treatment of Alzheimer's disease. Sci Rep. 2017 Apr 12;7:46320.
82. Schaefer TL, Braun AA, Amos-Kroohs RM, Williams MT, Ostertag E, Vorhees CV. A new model of Pde4d deficiency: genetic knock-down of PDE4D enzyme in rats produces an antidepressant phenotype without spatial cognitive effects. Genes Brain Behav. 2012 Jul;11(5):614-22.
83. Wang ZZ, Yang WX, Zhang Y, Zhao N, Zhang YZ, Liu YQ, et al. Phosphodiesterase-4D Knock-down in the Prefrontal Cortex Alleviates Chronic Unpredictable Stress-Induced Depressive-Like Behaviors and Memory Deficits in Mice. Sci Rep. 2015 Jul 10;5:11332.
84. Wang ZZ, Zhang Y, Liu YQ, Zhao N, Zhang YZ, Yuan L, et al. RNA interference-mediated phosphodiesterase 4D splice variants knock-down in the prefrontal cortex produces antidepressant-like and cognition-enhancing effects. Br J Pharmacol. 2013 Feb;168(4):1001-14.
85. Zhang C, Cheng Y, Wang H, Wang C, Wilson SP, Xu J, et al. RNA interference-mediated knockdown of long-form phosphodiesterase-4D (PDE4D) enzyme reverses amyloid-β42-induced memory deficits in mice. J Alzheimers Dis. 2014;38(2):269-80.
86. Baumgärtel K, Green A, Hornberger D, Lapira J, Rex C, Wheeler DG, et al. PDE4D regulates Spine Plasticity and Memory in the Retrosplenial Cortex. Sci Rep. 2018 Mar 1;8(1):3895.
87. Paes D, Schepers M, Willems E, Rombaut B, Tiane A, Solomina Y, et al. Ablation of specific long PDE4D isoforms increases neurite elongation and conveys protection against amyloid-β pathology. Cell Mol Life Sci. 2023 Jun 12;80(7):178.
88. Shi Y, Lv J, Chen L, Luo G, Tao M, Pan J, et al. Phosphodiesterase-4D Knockdown in the Prefrontal Cortex Alleviates Memory Deficits and Synaptic Failure in Mouse Model of Alzheimer's Disease. Front Aging Neurosci. 2021 Sep 3;13:722580.
89. Bolger GB, Peden AH, Steele MR, MacKenzie C, McEwan DG, Wallace DA, et al. Attenuation of the activity of the cAMP-specific phosphodiesterase PDE4A5 by interaction with the immunophilin XAP2. J Biol Chem. 2003 Aug 29;278(35):33351-63.
90. Millar JK, Pickard BS, Mackie S, James R, Christie S, Buchanan SR, et al. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science. 2005 Nov 18;310(5751):1187-91.
91. Clapcote SJ, Lipina TV, Millar JK, Mackie S, Christie S, Ogawa F, et al. Neuron. 2007 May 3;54(3):387-402.
92. Millar JK, Mackie S, Clapcote SJ, Murdoch H, Pickard BS, Christie S, et al. Disrupted in schizophrenia 1 and phosphodiesterase 4B: towards an understanding of psychiatric illness. J Physiol. 2007 Oct 15;584(Pt 2):401-5.
93. Murdoch H, Mackie S, Collins DM, Hill EV, Bolger GB, Klussmann E, et al. Isoform-selective susceptibility of DISC1/phosphodiesterase-4 complexes to dissociation by elevated intracellular cAMP levels. J Neurosci. 2007;27(35):9513-24.
94. Hayashi-Takagi A, Takaki M, Graziane N, Seshadri S, Murdoch H, Dunlop AJ, et al. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci. 2010 Mar;13(3):327-32.
95. Bradshaw NJ, Soares DC, Carlyle BC, Ogawa F, Davidson-Smith H, Christie S, et al. PKA phosphorylation of NDE1 is DISC1/PDE4 dependent and modulates its interaction with LIS1 and NDEL1. J Neurosci. 2011 Jun 15;31(24):9043-54.
96. Wang Q, Charych EI, Pulito VL, Lee JB, Graziane NM, Crozier RA, et al. The psychiatric disease risk factors DISC1 and TNIK interact to regulate synapse composition and function. Mol Psychiatry. 2011 Oct;16(10):1006-23.
97. Bradshaw NJ, Porteous DJ. DISC1-binding proteins in neural development, signalling and schizophrenia. Neuropharmacology. 2012 Mar;62(3):1230-41.
98. Lipina TV, Wang M, Liu F, Roder JC. Synergistic interactions between PDE4B and GSK-3: DISC1 mutant mice. Neuropharmacology. 2012 Mar;62(3):1252-62.
99. Tanaka M, Ishizuka K, Nekooki-Machida Y, Endo R, Takashima N, Sasaki H, et al. Aggregation of scaffolding protein DISC1 dysregulates phosphodiesterase 4 in Huntington's disease. J Clin Invest. 2017 Apr 3;127(4):1438-50.
100. Jeong MH, Urquhart G, Lewis C, Chi Z, Jewell JL. Inhibition of phosphodiesterase 4D suppresses mTORC1 signaling and pancreatic cancer growth. JCI Insight. 2023 Jul 10;8(13):e158098.
101. Cedervall P, Aulabaugh A, Geoghegan KF, McLellan TJ, Pandit J. Engineered stabilization and structural analysis of the autoinhibited conformation of PDE4. Proc Natl Acad Sci U S A. 2015 Mar 24;112(12):E1414-22.
102. Beard MB, Olsen AE, Jones RE, Erdogan S, Houslay MD, Bolger GB. UCR1 and UCR2 domains unique to the cAMP-specific phosphodiesterase family form a discrete module via electrostatic interactions. J Biol Chem. 2000 Apr 7;275(14):10349-58.
103. Richter W, Conti M. Dimerization of the type 4 cAMP-specific phosphodiesterases is mediated by the upstream conserved regions (UCRs). J Biol Chem. 2002 Oct 25;277(43):40212-21.
104. Xie M, Blackman B, Scheitrum C, Mika D, Blanchard E, Lei T, et al. The upstream conserved regions (UCRs) mediate homo- and hetero-oligomerization of type 4 cyclic nucleotide phosphodiesterases (PDE4s). Biochem J. 2014 May 1;459(3):539-50.
105. Bolger GB, Dunlop AJ, Meng D, Day JP, Klussmann E, Baillie GS, et al. Dimerization of cAMP phosphodiesterase-4 (PDE4) in living cells requires interfaces located in both the UCR1 and catalytic unit domains. Cell Signal. 2015 Apr;27(4):756-69.
106. Sette C, Conti M. Phosphorylation and activation of a cAMP-specific phosphodiesterase by the cAMP-dependent protein kinase. Involvement of serine 54 in the enzyme activation. J Biol Chem. 1996 Jul 12;271(28):16526-34.
107. Hoffmann R, Wilkinson IR, McCallum JF, Engels P, Houslay MD. cAMP-specific phosphodiesterase HSPDE4D3 mutants which mimic activation and changes in rolipram inhibition triggered by protein kinase A phosphorylation of Ser-54: generation of a molecular model. Biochem J. 1998 Jul 1;333 ( Pt 1)(Pt 1):139-49.
108. Lynch DC, Dyment DA, Huang L, Nikkel SM, Lacombe D, Campeau PM, et al. Identification of novel mutations confirms PDE4D as a major gene causing acrodysostosis. Hum Mutat. 2013 Jan;34(1):97-102.
109. Gardner OFW, Bai T, Baillie GS, Ferretti P. Phosphodiesterase 4D activity in acrodysostosis-associated neural pathology: too much or too little? Brain Commun. 2024 Jun 29;6(4):fcae225.
110. Burgin AB, Magnusson OT, Singh J, Witte P, Staker BL, Bjornsson JM, et al. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat Biotechnol. 2010 Jan;28(1):63-70.
111. Sutcliffe JS, Beaumont V, Watson JM, Chew CS, Beconi M, Hutcheson DM, et al. Efficacy of selective PDE4D negative allosteric modulators in the object retrieval task in female cynomolgus monkeys (Macaca fascicularis). PLoS One. 2014 Jul 22;9(7):e102449.
112. Gurney ME, Cogram P, Deacon RM, Rex C, Tranfaglia M. Multiple Behavior Phenotypes of the Fragile-X Syndrome Mouse Model Respond to Chronic Inhibition of Phosphodiesterase-4D (PDE4D). Sci Rep. 2017 Nov 7;7(1):14653.
113. Cui SY, Yang MX, Zhang YH, Zheng V, Zhang HT, Gurney ME, et al. Protection from Amyloid β Peptide-Induced Memory, Biochemical, and Morphological Deficits by a Phosphodiesterase-4D Allosteric Inhibitor. J Pharmacol Exp Ther. 2019 Nov;371(2):250-9.
114. Gurney ME, Nugent RA, Mo X, Sindac JA, Hagen TJ, Fox D 3rd, et al. Design and Synthesis of Selective Phosphodiesterase 4D (PDE4D) Allosteric Inhibitors for the Treatment of Fragile X Syndrome and Other Brain Disorders. J Med Chem. 2019 May 23;62(10):4884-901.
115. Jino K, Miyamoto K, Kanbara T, Unemura C, Horiguchi N, Ago Y. Allosteric inhibition of phosphodiesterase 4D induces biphasic memory-enhancing effects associated with learning-activated signaling pathways. Psychopharmacology (Berl). 2024 Apr;241(4):805-16.
116. Omar F, Findlay JE, Carfray G, Allcock RW, Jiang Z, Moore C, et al. Small-molecule allosteric activators of PDE4 long form cyclic AMP phosphodiesterases. Proc Natl Acad Sci U S A. 2019 Jul 2;116(27):13320-9.
117. Baillie GS, Tejeda GS, Kelly MP. Therapeutic targeting of 3',5'-cyclic nucleotide phosphodiesterases: inhibition and beyond. Nat Rev Drug Discov. 2019 Oct;18(10):770-96.
118. Boichot E, Wallace JL, Germain N, Corbel M, Lugnier C, Lagente V, et al. Anti-inflammatory activities of a new series of selective phosphodiesterase 4 inhibitors derived from 9-benzyladenine. J Pharmacol Exp Ther. 2000 Feb;292(2):647-53.
119. Yougbare I, Morin C, Senouvo FY, Sirois C, Albadine R, Lugnier C, et al. NCS 613, a potent and specific PDE4 inhibitor, displays anti-inflammatory effects on human lung tissues. Am J Physiol Lung Cell Mol Physiol. 2011 Oct;301(4):L441-50.
120. Keravis T, Monneaux F, Yougbaré I, Gazi L, Bourguignon JJ, Muller S, et al. Disease progression in MRL/lpr lupus-prone mice is reduced by NCS 613, a specific cyclic nucleotide phosphodiesterase type 4 (PDE4) inhibitor. PLoS One. 2012;7(1):e28899.
121. Naganuma K, Omura A, Maekawara N, Saitoh M, Ohkawa N, Kubota T, et al. Discovery of selective PDE4B inhibitors. Bioorg Med Chem Lett. 2009 Jun 15;19(12):3174-6.
122. Tang L, Huang C, Zhong J, He J, Guo J, Liu M, et al. Discovery of arylbenzylamines as PDE4 inhibitors with potential neuroprotective effect. Eur J Med Chem. 2019 Apr 15;168:221-31.
123. Xia C, Wen H, Zheng L, Ni Y, Bi H, Wang H, et al. Discovery of 7-alkoxybenzofurans as PDE4 inhibitors with hepatoprotective activity in D-GalN/LPS-induced hepatic sepsis. Eur J Med Chem. 2024 Sep 5;275:116576.
124. Hagen TJ, Mo X, Burgin AB, Fox D 3rd, Zhang Z, Gurney ME. Discovery of triazines as selective PDE4B versus PDE4D inhibitors. Bioorg Med Chem Lett. 2014 Aug 15;24(16):4031-4.
125. Silverberg JI, French LE, Warren RB, Strober B, Kjøller K, Sommer MOA, et al. Pharmacology of orismilast, a potent and selective PDE4 inhibitor. J Eur Acad Dermatol Venereol. 2023 Apr;37(4):721-9.
126. Burkovetskaya ME, Liu Q, Vadukoot AK, Gautam N, Alnouti Y, Kumar S, et al. KVA-D-88, a Novel Preferable Phosphodiesterase 4B Inhibitor, Decreases Cocaine-Mediated Reward Properties in Vivo. ACS Chem Neurosci. 2020 Aug 5;11(15):2231-42.
127. Herrmann FE, Hesslinger C, Wollin L, Nickolaus P. BI 1015550 is a PDE4B Inhibitor and a Clinical Drug Candidate for the Oral Treatment of Idiopathic Pulmonary Fibrosis. Front Pharmacol. 2022 Apr 20;13:838449. doi: 10.3389/fphar.2022.838449. Erratum in: Front Pharmacol. 2023 May 30;14:1219760.