Abstract
Despite a young age and limited exposure to nutritional challenges and other diseases, young children can develop liver disorders such as fatty liver, fibrosis, and liver cancer. The preliminary stages of these disorders are silent, and the patients come to the clinic at a late stage. During studies of pediatric liver cancer hepatoblastoma (HBL), our group found that a portion of our fresh HBL biobank is characterized by the development of Non-Alcoholic Fatty Liver Disease (NAFLD) including fatty liver, fibrosis, and increased proliferation. Our studies and other recent reports revealed that liver cancer and these fatty liver-specific disorders are strongly correlated with epigenetic changes in gene expression independent of HBL mutations. One of the mutation-independent pathways we focused on is the phosphorylation/de-phosphorylation status of C/EBPα at Ser190 (Ser193 in the mouse C/EBPα protein). De-phosphorylated C/EBPα loses its ability to interact with the chromatin remodeling protein p300, reducing transcription of the genes containing C/EBPα binding sites in their promoters. We found that the lack of the S193 phosphorylation of C/EBPα in S193A mice prevents the development of NAFLD in mouse models of pre-natal and post-natal high-fat diet-mediated NAFLD [1]. In this review, we present extended studies of NAFLD in both mouse models and HBL patients, which revealed the critical role of proliferation in the development of pediatric fatty liver disorders. Since Ser190 of C/EBPα is phosphorylated by cyclin D1-dependent kinase CDK4, our findings provided a rationale for the consideration of CDK4 inhibitors as potential drugs for the prevention of NAFLD and liver cancer in young kids. This review summarizes our findings and available data from other groups demonstrating the critical role of C/EBPα-dependent epigenetic pathways in the development of liver disorders in young children.
Keywords
Maternal obesity, Fatty liver, Fibrosis, Liver proliferation, C/EBPα
Abbreviations:
C/EBPα: CCAAT Enhancer Binding Protein alpha; HFD: High Fat Diet; HBL: Hepatoblastoma; HCC: Hepatocellular Carcinoma; NAFLD: Non-Alcoholic Fatty Liver Disease; OWOB: Overweight or Obese; cdk4: cyclin dependent kinase 4
Development of NAFLD in Young Children and Mouse Models under Pre-Natal High-Fat Diet Methods
Non-Alcoholic Fatty Liver Disease (NAFLD) in adult patients is the second highest cause of liver resections [2,3]. Fatty liver disorders in pediatric patients are underdiagnosed due to no or mild clinical symptoms at the early stages of liver disease. Considerable progress has been made in recent years by investigations of large cohorts of young patients. These studies have found a strong correlation between overweight or obese (OWOB) mothers during pregnancy and the subsequent development of liver disorders in their children [4-8]. Therefore, elucidating the molecular mechanisms that are involved in NAFLD caused by a high-fat-mediated diet is highly significant. A growing number of studies in animal models have shown that maternal fat intake is correlated with the development of a fatty liver phenotype and liver disorders in offspring [9-11]. These studies also provide new insights into how the development of maternal obese-mediated NAFLD in offspring can be inhibited. Particularly, Thompson’s group has shown that maternal exercise protects male offspring from maternal diet-programmed NAFLD [12]. A detailed description of the studies of animal models of parental obesity can be found in a review by Dr. Thompson [13].
Phosphorylation of Ser190 in C/EBPα Controls Its Ability to Promote/Inhibit the Development of NAFLD and Liver Cancer in Adults
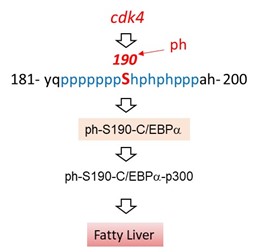
Figure 1. The critical role of phosphorylation of C/EBPα at Ser190 in the development of fatty liver in adults.
Regulation of C/EBPα Activities in Livers of Pediatric Patients
Most early studies were focused on the investigations of C/EBPα functions in the livers of healthy adult patients and liver disorders in adulthood. During the last decade, several groups began examining the prenatal and early postnatal activities of C/EBPα in both children and mouse models. Particularly, our lab has examined C/EBPα expression and post-translational modifications in the livers of young children with hepatoblastoma. These studies have unveiled that two groups of HBL patients have opposite expressions of C/EBPα mRNA. One group of HBL patients has reduced C/EBPα mRNA and protein due to the transcriptional repression and Gankyrin-mediated degradation of the C/EBPα protein [21]. The second group of HBL patients has elevated C/EBPα mRNA and C/EBPα protein [1,22,23]. Contrary to the tumor-suppressing activities of C/EBPα in adult livers, the elevation of ph-S190-C/EBPα in young children leads to the promotion of liver cancer [1,23]. In support of these observations, a recent study revealed that C/EBPα participates in a caffeine-dependent development of fatty liver phenotype in the offspring of maternal obesity settings [24].
Inhibition of Epigenetic Pathways Mediated by C/EBPα in S193A Mice Blocks the Development of Liver Disorders in High-Fat Diet Pregnancy-Associated Fatty Liver and NASH
Given the identification of new biological activities of C/EBPα in young children, we examined the role of the ph-S193/190-C/EBPα pathway in the development of high-fat diet pregnancy-associated liver disorders in offspring using a genetically modified mouse model C/EBPα-S193A. In these mice, C/EBPα cannot be phosphorylated at Ser193 and cannot form complexes with p300. Therefore, C/EBPα-p300-dependent pathways do not operate in these mice. Four arms were created for our study to compare the effects of a regular and high-fat diet on pups in utero. Wildtype and S193A-C/EBPα female mice were kept on either a normal or high-fat diet for six weeks before mating, and during pregnancy. Pups were weaned 28 days later and stratified into either a normal or high-fat diet. Reflecting the healthy diet was the normal-normal (NN) arm. The normal-high (NH) were mice whose mothers were on the normal diet during pregnancy and weaned to an HFD. The high-normal (HN) arm was mice whose mothers were exposed to an HFD during pregnancy and weaned to a normal diet. Finally, the high-high (HH) arm was mice whose mothers were on an HFD during pregnancy and weaned to an HFD.
Figure 2 summarizes results of the studies of HH arms of WT mice and C/EBPα-S193A mice. The HH arm of WT mice recapitulates the human condition where children of overweight mothers were fed high-fat food after birth. The animals were sacrificed 16 weeks after birth, and their livers were examined with a focus on liver disorders–fatty liver, fibrosis, liver proliferation, and NASH [1]. We found that the mutation of Ser193 to Ala changes the biological activities of C/EBPα by altering protein-protein interactions and blocking the development of fatty liver, fibrosis, NASH, and cell proliferation in the offspring. In contrast, these disorders are observed in the livers of WT mice, prenatally treated with a high-fat diet (Figure 2) [1]. These studies revealed the critical role of phosphorylation of C/EBPα in the development of liver disorders mediated by obesity and by a high-fat diet. The underlying mechanisms that blocked the development of fatty liver, fibrosis, and NASH within C/EBPα-S193A mice are associated with the inability of the mutant C/EBPα-S193A to interact with p300 and active genes involved in the development of the aforementioned disorders. Since CDK4 phosphorylates Ser193/Ser190 of C/EBPα, our studies suggest that CDK4 inhibitors might be used as potential drugs for liver disorders in young children. Additional support for the consideration of CDK4 as a therapeutic target was obtained in our extended studies of WT maternal high-fat diet-treated mice and in studies of HBL samples with fatty livers, described below.

Figure 2. A diagram showing the design for HH arms of WT mice and C/EBPα-S193A mice and the main results. See text for more details.
The Non-Fatty Liver Regions in Offspring of High-Fat Diet-Treated WT Mice and Non-Fatty Liver Regions in HBL Patients Show a High Rate of Proliferation Compared to Fatty Liver Regions
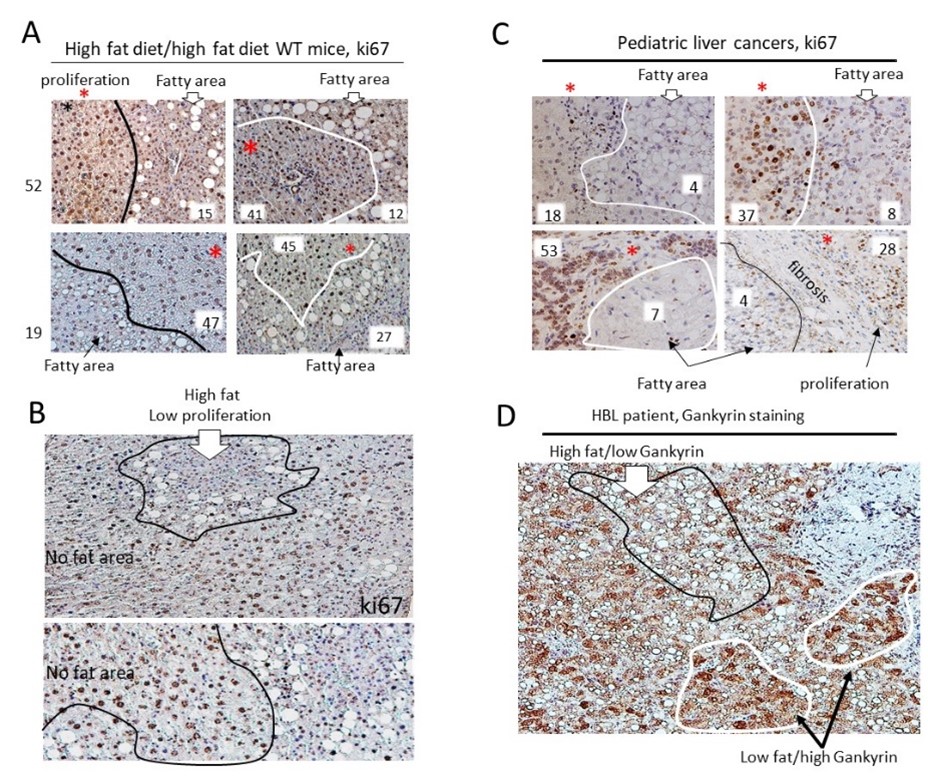
Figure 3. Translational aspects of the studies of offspring of pregnancy-obese mice related to liver disorders observed in obese children. A) Identification of fatty liver regions in the HH arm of WT mice with low levels of proliferation. Typical images of ki67 staining of the HH arm of WT mice are shown. Regions with a low number of fat droplets have higher rates of proliferation. Numbers of ki67-positive hepatocytes are shown in each area. B) Images show examples of large fields of HH arm of livers in WT mice with differential distribution of fat droplets and ki67-positive hepatocytes. C) Examples of regions of liver tumors in HBL patients with high fat/low proliferation and low fat/high proliferation. D) Staining of a fatty HBL liver to Gankyrin, a marker of liver cancer. Regions with high fat show low proliferation while low-fat regions have high proliferation, suggesting that fat droplets might be a protective response against cancer development. The experimental procedures of the staining can be found in our original paper [1].
As shown in Figure 3D, Gankyrin signals are strong in fatty HBL tumor regions with no or low amounts of fat droplets, while Gankyrin signals are weak in areas with abundant fat droplets. These findings suggest that increased proliferation is one of the key pathological events in the development of liver disorders in children.
Although these new findings are correlative, together with molecular studies in mouse models [1], they provide a rationale for considering therapeutic approaches targeting proliferation pathways, rather than pathways of developing fatty liver. One of the specific proliferation targets is CDK4 since this kinase directly phosphorylates C/EBPα at Ser193/190 and activates the C/EBPα-p300 pathway leading to liver disorders in offspring [1]. In this regard, our studies of CDK4 inhibitors (which drive liver proliferation) in adult livers showed a promising effect on inhibiting several features of NAFLD, such as fibrosis and NASH [17]. In agreement with these findings, it has been shown that cell cycle drivers CDK4 and E2F1 have additional activities that are associated with the regulation of lipogenesis, metabolism, and hepatic steatosis [28-30]. Given these data and the fact that livers in young children still proliferate, our studies provide a strong rationale to consider inhibitors of liver proliferation as potential drugs for the prevention of prenatal-obese-mediated fatty liver disorders.
Conflict of Interst
The authors have no conflict of interst.
Funding
The work was supported by internal funds from the Division of Pediatric Surgery (CCHMC), by NIH R01 CA278834 (NT), and by funds from Grace Foundation.
References
2. Wegermann K, Suzuki A, Mavis AM, Abdelmalek MF, Diehl AM, Moylan CA. Tackling nonalcoholic fatty liver disease: three targeted populations. Hepatology. 2021 Mar 1;73(3):1199-206.
3. Cantoral A, Montoya A, Luna‐Villa L, Roldán‐Valadez EA, Hernández‐Ávila M, Kershenobich D, et al. Overweight and obesity status from the prenatal period to adolescence and its association with non‐alcoholic fatty liver disease in young adults: cohort study. BJOG: An International Journal of Obstetrics & Gynaecology. 2020 Sep;127(10):1200-9.
4. Mosca A, De Cosmi V, Parazzini F, Raponi M, Alisi A, Agostoni C, et al. The role of genetic predisposition, programing during fetal life, family conditions, and post-natal diet in the development of pediatric fatty liver disease. The Journal of pediatrics. 2019 Aug 1;211:72-7.
5. Stratakis N, Conti DV, Jin R, Margetaki K, Valvi D, Siskos AP, et al. Prenatal exposure to perfluoroalkyl substances associated with increased susceptibility to liver injury in children. Hepatology. 2020 Nov 1;72(5):1758-70.
6. Ramírez-Mejía MM, Díaz-Orozco LE, Barranco-Fragoso B, Méndez-Sánchez N. A review of the increasing prevalence of metabolic-associated fatty liver disease (MAFLD) in children and adolescents worldwide and in Mexico and the implications for public health. Medical Science Monitor: International Medical Journal of Experimental and Clinical Research. 2021;27:e934134.
7. Félix DR, Costenaro F, Gottschall CB, Coral GP. Non-alcoholic fatty liver disease (Nafld) in obese children-effect of refined carbohydrates in diet. BMC Pediatrics. 2016 Dec;16:187.
8. De Jesus DF, Orime K, Kaminska D, Kimura T, Basile G, Wang CH, et al. Parental metabolic syndrome epigenetically reprograms offspring hepatic lipid metabolism in mice. The Journal of Clinical Investigation. 2020 May 27;130(5):2391-407.
9. Alfaradhi MZ, Fernandez-Twinn DS, Martin-Gronert MS, Musial B, Fowden A, Ozanne SE. Oxidative stress and altered lipid homeostasis in the programming of offspring fatty liver by maternal obesity. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2014 Jul 1;307(1):R26-34.
10. Thompson MD, Cismowski MJ, Trask AJ, Lallier SW, Graf AE, Rogers LK, et al. Enhanced steatosis and fibrosis in liver of adult offspring exposed to maternal high-fat diet. Gene Expression. 2016;17(1):47-59.
11. Thompson MD, Derse A, Ferey JL, Reid M, Xie Y, Christ M, et al. Transgenerational impact of maternal obesogenic diet on offspring bile acid homeostasis and nonalcoholic fatty liver disease. American Journal of Physiology-Endocrinology and Metabolism. 2019 Apr 1;316(4):E674-86.
12. Hinrichs H, Faerber A, Young M, Ballentine SJ, Thompson MD. Maternal Exercise Protects Male Offspring From Maternal Diet–Programmed Nonalcoholic Fatty Liver Disease Progression. Endocrinology. 2023 Mar 1;164(3):bqad010.
13. Thompson MD. Developmental programming of NAFLD by parental obesity. Hepatology Communications. 2020 Oct;4(10):1392-403.
14. Cao Z, Umek RM, McKnight SL. Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes & Development. 1991 Sep 1;5(9):1538-52.
15. Lourenço AR, Coffer PJ. A tumor suppressor role for C/EBPα in solid tumors: more than fat and blood. Oncogene. 2017 Sep;36(37):5221-30.
16. Timchenko NA. Aging and liver regeneration. Trends in Endocrinology & Metabolism. 2009 May 1;20(4):171-6.
17. Nguyen P, Valanejad L, Cast A, Wright M, Garcia JM, El-Serag HB, et al. Elimination of age-associated hepatic steatosis and correction of aging phenotype by inhibition of cdk4-C/EBPα-p300 axis. Cell Reports. 2018 Aug 7;24(6):1597-609.
18. Jin J, Iakova P, Breaux M, Sullivan E, Jawanmardi N, Chen D, et al. Increased expression of enzymes of triglyceride synthesis is essential for the development of hepatic steatosis. Cell Reports. 2013 Mar 28;3(3):831-43.
19. Jin J, Valanejad L, Nguyen TP, Lewis K, Wright M, Cast A, et al. Activation of CDK4 triggers development of non-alcoholic fatty liver disease. Cell Reports. 2016 Jul 19;16(3):744-56.
20. Cast A, Valanejad L, Wright M, Nguyen P, Gupta A, Zhu L, et al. C/EBPα‐dependent preneoplastic tumor foci are the origin of hepatocellular carcinoma and aggressive pediatric liver cancer. Hepatology. 2018 May;67(5):1857-71.
21. Valanejad L, Lewis K, Wright M, Jiang Y, D’Souza A, Karns R, et al. FXR-Gankyrin axis is involved in development of pediatric liver cancer. Carcinogenesis. 2017 Jul 1;38(7):738-47.
22. Valanejad L, Cast A, Wright M, Bissig KD, Karns R, Weirauch MT, et al. PARP1 activation increases expression of modified tumor suppressors and pathways underlying development of aggressive hepatoblastoma. Communications Biology. 2018 Jun 11;1(1):67.
23. Rivas M, Johnston II ME, Gulati R, Kumbaji M, Aguiar TF, Timchenko L, et al. HDAC1-dependent repression of markers of hepatocytes and P21 is involved in development of pediatric liver cancer. Cellular and Molecular Gastroenterology and Hepatology. 2021 Jan 1;12(5):1669-82.
24. Hu S, Xia L, Luo H, Xu Y, Yu H, Xu D, et al. Prenatal caffeine exposure increases the susceptibility to non-alcoholic fatty liver disease in female offspring rats via activation of GR-C/EBPα-SIRT1 pathway. Toxicology. 2019 Apr 1;417:23-34.
25. Cast A, Kumbaji M, D'Souza A, Rodriguez K, Gupta A, Karns R, et al. Liver proliferation is an essential driver of fibrosis in mouse models of nonalcoholic fatty liver disease. Hepatology Communications. 2019 Aug;3(8):1036-49.
26. Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology. 2007 Jun;45(6):1366-74.
27. Sakurai T, Yada N, Hagiwara S, Arizumi T, Minaga K, Kamata K, et al. Gankyrin induces STAT 3 activation in tumor microenvironment and sorafenib resistance in hepatocellular carcinoma. Cancer Science. 2017 Oct;108(10):1996-2003.
28. Denechaud PD, Lopez-Mejia IC, Fajas L. Cell cycle regulators CDK4 and E2F1 control glucose and lipid homeostasis. Medecine Sciences: M/S. 2016 Oct;32(10):815-8.
29. Denechaud PD, Lopez-Mejia IC, Giralt A, Lai Q, Blanchet E, Delacuisine B, et al. E2F1 mediates sustained lipogenesis and contributes to hepatic steatosis. The Journal of Clinical Investigation. 2016 Jan 4;126(1):137-50.
30. Denechaud PD, Fajas L, Giralt A. E2F1, a novel regulator of metabolism. Frontiers in endocrinology. 2017 Nov 10;8:311.