Commentary
Cardiovascular disease (CVD) is a leading cause of maternal mortality worldwide [1,2]. The molecular mechanisms underlying de novo CVD in pregnancy are not fully understood, which has limited targeted therapeutic development. Our group’s recent article “Placental senescence pathophysiology is shared between peripartum cardiomyopathy and preeclampsia in mouse and human” [3], provides new insights into the pathobiology of pregnancy-related CVD. Specifically, we studied two closely related CVDs in pregnancy, peripartum cardiomyopathy (PPCM), an idiopathic form of heart failure (HF) that occurs in late pregnancy to early postpartum, and preeclampsia, a common hypertensive disorder of pregnancy that is a major risk factor for postpartum HF. Using a serum proteomics approach, we discovered a fundamental aging process, known as senescence, as one of the most highly dysregulated biological processes in pregnant patients with PPCM or preeclampsia. Our work further identified the placenta as the likely primary source of circulating proteins in the maternal blood contributing to the senescence-associated secretory phenotype (SASP). We identified 28 SASP proteins shared between PPCM and preeclampsia and found that placental-derived SASP (i.e. activin A) correlates with cardiac dysfunction and HF severity in patients with PPCM or preeclampsia. In a mouse model of PPCM, the cardiac specific PGC1a knockout mouse [4], we showed that restoring placental senescence back to homeostatic physiological levels with the senolytic, fisetin, can improve cardiac function and adverse myocardial remodeling in PPCM. Directly inhibiting placental-derived SASP, e.g. activin A, in the early postpartum period similarly improved cardiac function in this mouse model of PPCM. Overall, the findings from this study lead to an emerging concept that dysregulated placental senescence and its associated SASP may be part of the core pathophysiology driving CVD in pregnancy. However, many questions remain about how this normal physiological process becomes dysregulated in pregnancy and the mechanisms by which the placenta crosstalks with the maternal heart. This commentary discusses data from our published work that provides additional insights into the origin of increased placental senescence and SASP in pregnancy that can contribute to the transition between health and disease in pregnancy.
Placental Senescence
To understand the pathological implications of placental senescence, it is important to recognize that cellular senescence is part of the normal physiology of pregnancy. Senescence plays a critical role in the temporal and spatial regulation of fetal development, notably through the senescence-associated secretory phenotype (SASP), a composite of all the secreted factors generated by senescent cells [5,6]. It is essential for regulating placental development and function [7,8]. In the placenta, the features of senescence are induced by DNA damage and involves the coordinated activation of the p53/p21 and p16/pRb regulatory pathways [9].
Senescence pathways participate in placental development and function throughout pregnancy and serve distinct roles in different placental zones and cell types [7,8]. Notably, cytotrophoblasts exhibit high expression of senescence markers, such as senescence-associated β-galactosidase (SA-β-gal), p16, and p21, which decline during the third trimester. In contrast, syncytiotrophoblasts (SCT) display lower levels of senescence in the first and second trimesters, but this progressively increases throughout pregnancy, peaking in the third trimester [10]. The cell fusion that leads to the formation of the SCT is strongly associated with senescence [9]. This helps maintain the SCT’s non-proliferative state, preserves its viability, and regulates interactions with the immune system and tissue remodeling through SASP.
The precise regulation of placental senescence plays a vital role in facilitating successful placental implantation, delivery, and pregnancy outcomes [11-14]. For example, decreases in physiological placental senescence are linked to implantation failure and fetal growth restrictions, while excessive senescence is associated with recurrent miscarriage [15-17]. Ultimately, both increases and decreases in placental senescence can contribute to adverse pregnancy outcomes. Thus, understanding the temporal, spatial, and cell-specific properties that govern this aging process in the placenta is critical in the development of therapeutics that could modulate these pathways.
Insight from Placental Histology and Immunostaining
Placental senescence localization during healthy pregnancy
In our study, SA-β-gal staining in pregnant C57BL/6 mice confirmed a progressive increase in placental senescence throughout gestation, consistent with previous findings [10,15]. Notably, our analysis also identified regional- and cell-specific differences in placental senescence in the contexts of health and disease.
Our study analyzed placentas from both mice and humans. While mammalian placentas exhibit functional convergence, their morphology does vary across species. The murine placenta can be divided into three distinct zones: the decidua, the labyrinth, and the area between them: the junctional zone [18]. In humans, the structure analogous to the murine labyrinth is the placental villi [19]. The decidua is the maternal portion of the placenta, originating from the uterine endometrium. This region contains decidual stromal cells and extravillous trophoblasts. The labyrinth, equivalent to the chorionic villi in the human placenta, is a fetal-side structure that serves as the primary maternal-fetal exchange zone. This zone is rich in SCT, cytotrophoblasts (CT), and vascular cells [20,21]. Finally, the junctional zone corresponds to the transitional region between the decidua and the labyrinth and is composed of three cell types: spongiotrophoblast cells (SpTs), glycogen cells (GCs), and trophoblast giant cells (TGCs).
In term placentas from normal murine pregnancy, we found a gradient of SA-β-gal staining across the placental zones, with the strongest expression in the decidua and the weakest in the labyrinth. These results are consistent with prior studies, which have similarly shown an increase in senescence in the decidua with increased p21, SASP (e.g. IL-6 and IL-8), and SA-β-gal levels, along with decreased telomere length [12-14]. In parallel with SA-β-gal staining, we also performed comprehensive immunostaining of cell-lineage markers for trophoblasts (cytokeratin 7), stromal cells (vimentin), endothelial cells (CD31), and multiple immune cell populations. These experiments demonstrated colocalization of senescence with stromal cells located predominantly in the decidua and less in the junctional zone. Senescent trophoblasts were mainly found in the junctional zone. Trophoblasts are predominantly located in the labyrinth where the senescence signal is substantially lower (Figure 1). Immunostaining of placental cotyledon samples from patients with normal pregnancy similarly demonstrated colocalization of senescence markers (SA-β-gal, p21, activin A) with trophoblasts. Notably, the decidua was not included in our human placental specimens, limiting conclusions on the maternal stromal contribution. In summary, our study showed that placental senescence progressively increases during pregnancy and stems, in part, from stromal and trophoblastic cell populations.
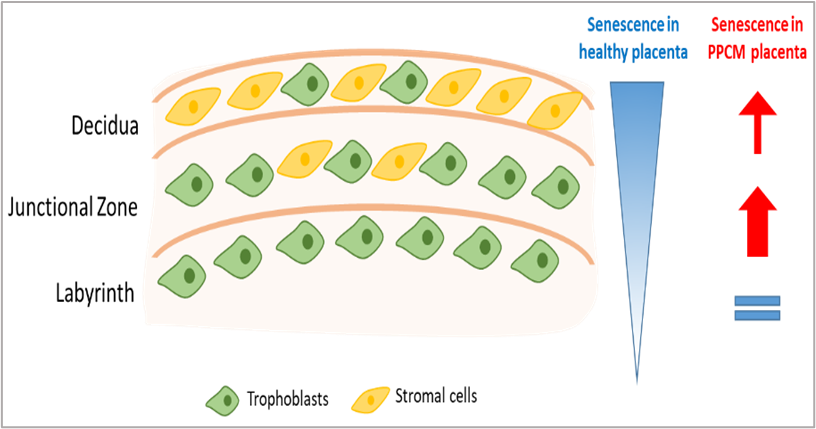
Figure 1. Schematic of regional- and cell-specific differences in placental senescence in control versus PPCM mice at gestational day 17. Proportions of trophoblasts and stromal cells according to placental zones (decidua, junctional zone, and labyrinth), and levels of senescence (white = low, blue = high) in the three zones under healthy conditions and in PPCM mice.
Placental senescence localization in PPCM and preeclampsia
Although our study primarily assessed changes in global placental senescence in disease states (i.e. PPCM or preeclampsia), our analyses do provide some initial insights into the spatial- and cell-specific changes in placental senescence in these pathologic contexts. In our PPCM mouse model, we observed an increase in overall placental senescence, which was mostly due to an increase in senescence in the junctional zone and a trend in the decidua, while the labyrinth remained unchanged (Figure 1). Notably, this increase in senescence colocalized with stromal and trophoblast cell populations. In patients with preeclampsia, the increase in senescence markers primarily colocalized with trophoblasts, but again, our analysis did not include the decidua. Ultimately, although limited in scope, our findings highlight the stromal and trophoblast populations as potential origins of the pathological increase of placental senescence observed in PPCM and preeclampsia.
Insights from Placental Single Cell and Single Nuclei RNA Sequencing
In addition to comprehensive immunostaining, we also performed targeted analysis of single cell (sc) and single nuclei (sn) RNAseq data from placentas of pregnancies with early preeclampsia compared to uncomplicated pregnancies with similar timing of delivery [22]. Our analysis specifically focused on hallmark senescence genes (CDKN1A, CDKN2A) and the genes encoding the 28 SASP identified in our serum proteomics. Within these samples, 46 cell types were identified, which were clustered into four major groups: immune, stromal, trophoblast, and vascular cells. Of note, this analysis was done in placental cotyledon samples, in which the maternal decidua was removed at the time of sample collection.
Despite this limitation, some preliminary insights into the cell specificity of placental senescence can be made. In the placental samples from control subjects, i.e. individuals with preterm delivery but otherwise uncomplicated pregnancy, multiple SASP were expressed relatively uniformly across the four major cell groups, including PPBP, THBS2, CCL17, IL7, MMP7, CCL22, CCL11, and CXCL16. Interestingly, these were also not altered in the context of preeclampsia. Other SASP showed more cell-specific expression patterns, albeit with some overlap. ILRL1, DKK1, RARRES2, TIMP1, MRC2, CDKN1A, CDKN2A, TFPI, VEGFA, PDGFA, MAP2K2, SERPINE1, SCT1, PLAT, and FSTL3 were all highly expressed in stromal populations. FSTL3, SERPINE1, PDGFA, TFPI, and INHBA were expressed in trophoblasts, EREG, INHBA, MRC2, TIMP1, VEGFA, PDGFB, CCL18 in immune cells, and MMP1, MRC2, SELP, TIMP1, TFPI, RARRES2, VEGFA, PDGFA, PGDFB, CXCL12, PLAT, SERPINE1, FSTL3 in vascular cells.
Trophoblasts
Of the 28 SASP shared between PPCM and preeclampsia, we identified activin A as the most highly upregulated in human preeclamptic placenta. Activin A, encoded by the INHBA gene, and SASP associated with its signaling pathway—SERPINE1 and FSTL3—are all predominantly increased in trophoblasts (Table 1). This trend was also observed for TIMP1, TFPI, VEGFA, PDGFA, PDGFB, and MAP2K2. The increased expression of these SASP genes is present in extravillous trophoblasts (EVT), SCT, and villous cytotrophoblasts (VCT), with the majority observed in VCT. VCTs and SCTs constitute placental villi, while VCTs also form the cytotrophoblastic shell at the maternal-fetal interface, corresponding to the junctional zone and labyrinth. These results provide additional evidence supporting a role for trophoblast senescence in the placental senescence pathophysiology of preeclampsia and PPCM.
Indeed, prior work has identified an increase in trophoblast senescence in preeclampsia [23]. How this links to maternal preeclampsia phenotypes is not entirely clear, but based on our data and others, this is presumably mediated through increased trophoblast SASP production. Soluble Fms-like tyrosine kinase 1 (sFlt1), a secreted VEGF inhibitor, is produced by placental trophoblasts and contributes to the pathophysiology of both preeclampsia and PPCM [24-26]. Although sFlt1 is not a bona fide SASP factor, it represents strong evidence that secreted factors from trophoblast populations are causally involved in these diseases. Notably, similar drivers, including hypoxia and oxidative stress, induce both sFlt1 production and senescence in trophoblasts, suggesting that either similar or parallel pathology is likely occurring [27-31].
Stromal cells
While our study did not extensively characterize non-trophoblastic cells, our scRNAseq analysis showed that SASP genes, including SERPINE1, TIMP1, EREG, VEGFA, and DKK1, are also highly expressed in stromal cells (Table 1). Interestingly, recent work has shown that increased placental mesenchymal stem/stromal cell senescence can affect trophoblast and endothelial cell function in preeclampsia [32]. Similar SERPINE and TIMP-related genes are implicated in stromal cell senescence, in addition to DUSP1, IFIT2, and CTSZ [32]. Although the decidua was not analyzed in our scRNAseq analysis, given the marked increase in decidual stroma senescence in the mouse, we would propose that further investigations into senescence biology in these placental cell types and regions could yield new insights into the (patho)physiology of human pregnancy and disease.
Vascular cells
In vascular cells (Table 1), the increase in the 28 SASP identified in our study was most prominent in pericytes (TIMP1, TFPI), smooth muscle cells (TIMP1), and endothelial cells (SERPINE1, VEGFA, FSTL3). Endothelial dysfunction and impaired angiogenesis are part of the central pathophysiology of preeclampsia [33–35] and PPCM [4,30] and it is possible that endothelial senescence could also be part of this process. Pericytes also participate in angiogenesis and are critical in maintaining the functional microvascular network in the placenta [36]. Loss of pericyte coverage in the placental microvasculature has been shown to contribute to preeclampsia in mouse models [37]. However, whether this is related to pericyte senescence is unclear.
Immune cells
Lastly, multiple immune cells showed increased SASP expression (Table 1), particularly macrophages and Hofbauer cells, which display elevated levels of INHBA, TIMP1, and VEGFA. Hofbauer cells are placental villous macrophages of fetal origin that play roles in placental development, including vasculogenesis and angiogenesis. These cells participate in the immune imbalance leading to the pro-inflammatory state in preeclampsia [38,39]. While it is well established that activation of macrophage populations contributes to preeclampsia, how senescence potentially contributes to this process is unknown.
|
Placental cell types |
Increased placental SASPs and senescence genes in preeclampsia |
|
Stromal cells |
TIMP1, CDKN1A, CDKN2A, EREG, VEGFA, DKK1, SERPINE1, TFPI, EREG |
|
Trophoblasts |
INHBA, SERPINE1, FSTL3, CDKN2A, CDKN1A, TIMP1, TFPI, VEGFA, PDGFA, PDGFB, MAP2K2 |
|
Vascular cells |
VEGFA, CDKN1A, TIMP1, TFPI, FSTL3, STC1, SERPINE1 |
|
Immune cells |
INHBA, TIMP1, VEGFA |
Limitations
We would emphasize that our original paper was not a priori designed to fully characterize the cellular, temporal, and spatial regulation of senescence in the placenta. While some conclusions can be made from our placental characterization in the various mouse and human cohorts used, these should be viewed as preliminary within the limitations of the study. First, in the human studies, control subjects were mostly individuals with an uncomplicated pregnancy but preterm delivery. While this enabled us to match controls to preeclampsia patients, based on gestational age (an important consideration in assessing placental senescence), these placental samples do not fully reflect the dynamics of senescence in term pregnancies. Recent work, however, has shown that multiple placental SASP factors identified in preterm delivery are also upregulated in term pregnancy [40]. Second, the decidua was not included in our analyses of human placental samples, and thus, it is unclear if the senescence findings in the decidua of murine placental samples also occurs in humans. Lastly, the interventions on placental senescence were only performed in a mouse model of PPCM, which recapitulates some, but not all, of the features of clinical PPCM.
Summary
The placenta is a unique hybrid maternal-fetal organ that not only serves as a critical regulator of fetal health and development, but can also significantly impact maternal health via its dynamic secretome. Our recent paper, along with other’s work [4,41,42], strongly suggests that the crosstalk between the placenta and the maternal cardiovascular system plays a major role in the development and progression of CVD in pregnancy. Understanding how physiological placental aging can transition into a pathologic process and shift the balance between maternal health and disease is a critical first step before attempting to therapeutically intervene on this process.
Our findings identify senescent trophoblasts and stromal cells as some of the major players in this process. Why these cells become excessively senescent remains to be determined, but likely is multifactorial with contributions from oncogene activation [43], telomere shortening [44], oxidative stress [31], hypoxia [45], and other stressors that induce DNA damage [46]. The entire spectrum of placental-derived SASP that could affect the maternal cardiovascular system also is not clear. Ultimately, while our study provides some of the first data functionally linking placental senescence to maternal CVD, we are in the early stages of understanding the physiological and pathological implications of placental senescence in maternal and fetal health. Elucidating the molecular mechanisms governing the temporal, spatial, and cell-specific properties of placental senescence would not only advance our understanding of pregnancy-related CVD but could also pave the path for novel therapeutic strategies for these diseases.
References
2. Mehta LS, Warnes CA, Bradley E, Burton T, Economy K, Mehran R, et al; American Heart Association Council on Clinical Cardiology; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular and Stroke Nursing; and Stroke Council. Cardiovascular Considerations in Caring for Pregnant Patients: A Scientific Statement From the American Heart Association. Circulation. 2020 Jun 9;141(23):e884-e903.
3. Roh JD, Castro C, Yu A, Rana S, Shahul S, Gray KJ, et al; IPAC Investigators; Hilfiker-Kleiner D, Rosenzweig A. Placental senescence pathophysiology is shared between peripartum cardiomyopathy and preeclampsia in mouse and human. Sci Transl Med. 2024 Apr 17;16(743):eadi0077.
4. Patten IS, Rana S, Shahul S, Rowe GC, Jang C, Liu L, et al. Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature. 2012 May 9;485(7398):333-8.
5. Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, et al. Programmed cell senescence during mammalian embryonic development. Cell. 2013 Nov 21;155(5):1104-18.
6. Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013 Nov 21;155(5):1119-30.
7. Cox LS, Redman C. The role of cellular senescence in ageing of the placenta. Placenta. 2017 Apr;52:139-45.
8. Farfán-Labonne B, Leff-Gelman P, Pellón-Díaz G, Camacho-Arroyo I. Cellular senescence in normal and adverse pregnancy. Reprod Biol. 2023 Mar;23(1):100734.
9. Chuprin A, Gal H, Biron-Shental T, Biran A, Amiel A, Rozenblatt S, et al. Cell fusion induced by ERVWE1 or measles virus causes cellular senescence. Genes Dev. 2013 Nov 1;27(21):2356-66.
10. Higuchi S, Miyamoto T, Kobara H, Yamada S, Asaka R, Kikuchi N, et al. Trophoblast type-specific expression of senescence markers in the human placenta. Placenta. 2019 Sep 15;85:56-62.
11. Lai TP, Simpson M, Patel K, Verhulst S, Noh J, Roche N, et al. Telomeres and replicative cellular aging of the human placenta and chorioamniotic membranes. Sci Rep. 2021 Mar 4;11(1):5115.
12. Bonney EA, Krebs K, Saade G, Kechichian T, Trivedi J, Huaizhi Y, et al. Differential senescence in feto-maternal tissues during mouse pregnancy. Placenta. 2016 Jul;43:26-34.
13. Menon R, Behnia F, Polettini J, Saade GR, Campisi J, Velarde M. Placental membrane aging and HMGB1 signaling associated with human parturition. Aging (Albany NY). 2016 Feb 4;8(2):216-29.
14. Kajdy A, Modzelewski J, Cymbaluk-Płoska A, Kwiatkowska E, Bednarek-Jędrzejek M, Borowski D, et al. Molecular Pathways of Cellular Senescence and Placental Aging in Late Fetal Growth Restriction and Stillbirth. Int J Mol Sci. 2021 Apr 18;22(8):4186.
15. Gal H, Lysenko M, Stroganov S, Vadai E, Youssef SA, Tzadikevitch-Geffen K, et al. Molecular pathways of senescence regulate placental structure and function. EMBO J. 2019 Sep 16;38(18):e100849.
16. de la Torre P, Fernández-de la Torre M, Flores AI. Premature senescence of placental decidua cells as a possible cause of miscarriage produced by mycophenolic acid. J Biomed Sci. 2021 Jan 4;28(1):3.
17. Rawlings TM, Makwana K, Taylor DM, Molè MA, Fishwick KJ, Tryfonos M, et al. Modelling the impact of decidual senescence on embryo implantation in human endometrial assembloids. Elife. 2021 Sep 6;10:e69603.
18. Marsh B, Blelloch R. Single nuclei RNA-seq of mouse placental labyrinth development. Elife. 2020 Nov 3;9:e60266.
19. Woods L, Perez-Garcia V, Hemberger M. Regulation of Placental Development and Its Impact on Fetal Growth-New Insights From Mouse Models. Front Endocrinol (Lausanne). 2018 Sep 27;9:570.
20. Tai-Nagara I, Yoshikawa Y, Numata N, Ando T, Okabe K, Sugiura Y, et al. Placental labyrinth formation in mice requires endothelial FLRT2/UNC5B signaling. Development. 2017 Jul 1;144(13):2392-401.
21. Rinkenberger J, Werb Z. Nat Gen-2000-Reikenberger and Werb-The labyrinthe placenta. 2000;25:2-4.
22. Admati I, Skarbianskis N, Hochgerner H, Ophir O, Weiner Z, Yagel S, et al. Two distinct molecular faces of preeclampsia revealed by single-cell transcriptomics. Med. 2023 Oct 13;4(10):687-709.e7.
23. Sukenik-Halevy R, Amiel A, Kidron D, Liberman M, Ganor-Paz Y, Biron-Shental T. Telomere homeostasis in trophoblasts and in cord blood cells from pregnancies complicated with preeclampsia. Am J Obstet Gynecol. 2016 Feb;214(2):283.e1-283.e7.
24. Damp J, Givertz MM, Semigran M, Alharethi R, Ewald G, Felker GM, et al; IPAC Investigators. Relaxin-2 and Soluble Flt1 Levels in Peripartum Cardiomyopathy: Results of the Multicenter IPAC Study. JACC Heart Fail. 2016 May;4(5):380-8.
25. Davis MB, Arany Z, McNamara DM, Goland S, Elkayam U. Peripartum Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2020 Jan 21;75(2):207-21.
26. Damp JA, Arany Z, Fett JD, Blauwet L, Elkayam U. Imbalanced Angiogenesis in Peripartum Cardiomyopathy (PPCM). Circ J. 2018 Sep 25;82(10):2689.
27. Albers RE, Kaufman MR, Natale BV, Keoni C, Kulkarni-Datar K, Min S, et al. Trophoblast-Specific Expression of Hif-1α Results in Preeclampsia-Like Symptoms and Fetal Growth Restriction. Sci Rep. 2019 Feb 26;9(1):2742.
28. Grzeszczak K, Łanocha-Arendarczyk N, Malinowski W, Ziętek P, Kosik-Bogacka D. Oxidative Stress in Pregnancy. Biomolecules. 2023 Dec 9;13(12):1768.
29. Chiarello DI, Abad C, Rojas D, Toledo F, Vázquez CM, Mate A, et al. Oxidative stress: Normal pregnancy versus preeclampsia. Biochim Biophys Acta Mol Basis Dis. 2020 Feb 1;1866(2):165354.
30. Ricke-Hoch M, Pfeffer TJ, Hilfiker-Kleiner D. Peripartum cardiomyopathy: basic mechanisms and hope for new therapies. Cardiovasc Res. 2020 Mar 1;116(3):520-31.
31. Pole A, Dimri M, Dimri GP. Oxidative stress, cellular senescence and ageing. AIMS molecular science. 2016;3(3):300-24.
32. Zhang Y, Zhong Y, Yu Z, Cheng X, Zou L, Liu X. Single cell RNA-sequencing reveals the cellular senescence of placental mesenchymal stem/stromal cell in preeclampsia. Placenta. 2024 May;150:39-51.
33. Powe CE, Levine RJ, Karumanchi SA. Preeclampsia, a disease of the maternal endothelium: the role of antiangiogenic factors and implications for later cardiovascular disease. Circulation. 2011 Jun 21;123(24):2856-69.
34. Pabon MA, Weisbrod RM, Castro C, Li H, Xia P, Kang J, et al. Venous Endothelial Cell Transcriptomic Profiling Implicates METAP1 in Preeclampsia. Circ Res. 2025 Jan 17;136(2):180-90.
35. O'Brien M, Baczyk D, Kingdom JC. Endothelial Dysfunction in Severe Preeclampsia is Mediated by Soluble Factors, Rather than Extracellular Vesicles. Sci Rep. 2017 Jul 19;7(1):5887.
36. Harris SE, Matthews KS, Palaiologou E, Tashev SA, Lofthouse EM, Pearson-Farr J, et al. Pericytes on placental capillaries in terminal villi preferentially cover endothelial junctions in regions furthest away from the trophoblast. Placenta. 2021 Jan 15;104:1-7.
37. Morrison MJ, Natale BV, Allen S, Peterson N, Natale DRC. Characterizing placental pericytes: Hypoxia and proangiogenic signalling. Placenta. 2024 Sep 26;155:1-10.
38. Reyes L, Golos TG. Hofbauer Cells: Their Role in Healthy and Complicated Pregnancy. Front Immunol. 2018 Nov 15;9:2628.
39. Ma Y, Ye Y, Zhang J, Ruan CC, Gao PJ. Immune imbalance is associated with the development of preeclampsia. Medicine (Baltimore). 2019 Apr;98(14):e15080.
40. Barak O, Bauer AD, Parks WT, Lovelace TC, Benos PV, Chu T, et al. Characterization of senescence-associated transcripts in the human placenta. Placenta. 2025 Mar 6;161:31-38.
41. Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, et al. Circulating angiogenic factors and the risk of preeclampsia. N Engl J Med. 2004 Feb 12;350(7):672-83.
42. Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, et al . Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003 Mar;111(5):649-58.
43. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011 Feb 21;192(4):547-56.
44. Victorelli S, Passos JF. Telomeres and Cell Senescence - Size Matters Not. EBioMedicine. 2017 Jul;21:14-20.
45. Ciampa EJ, Flahardy P, Srinivasan H, Jacobs C, Tsai L, Karumanchi SA, et al. Hypoxia-inducible factor 1 signaling drives placental aging and can provoke preterm labor. Elife. 2023 Aug 23;12:RP85597.
46. Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014 Jul;15(7):482-96.