Abstract
Extracellular matrix proteins are produced by osteoblasts and chondrocytes in order to establish and sustain the skeletal system. As secretory cells, these cells must be endowed with a large capacity for protein synthesis, as well as the equipment for quality control and transport of freshly produced secreted proteins. To achieve this aim, we deem that the unfolded protein response (UPR) is important. Recent studies have revealed that the UPR plays a larger role in skeletal development than previously thought. The UPR is involved in many aspects of bone formation and homeostasis, as well as the development of skeletal diseases. A new analysis of UPR signaling molecules’ function in bone metabolism and disease biomarkers is presented in this article. Furthermore, we also analyzed the differential expression profile after overexpressing IRE1a and ATF6 in chondrocyte and discovered that ATF6 and IRE1a can directly interact with certain genes to participate in bone development and bone-related disease processes by transcription factor Chip-chip assay. These discoveries, based on the comprehensive analysis of Chip assay and literature reports, may predict the relationship between each UPR signaling molecule and bone metabolism. It is of far-reaching significance for the further study of the diagnosis and therapies strategies for skeletal diseases.
Keywords
UPR signaling molecules, ATF6, IRE1, XBP1, PERK, Bone formation and homeostasis, Osteoarthritis, Osteoporosis, Rheumatoid arthritis
Introduction
The skeletal system consists mostly of extracellular matrix and mineral salts. Osteoblasts and chondrocytes create enormous amounts of extracellular matrix proteins for skeletal systems to grow and sustain. Many skeletal metabolism related proteins are produced by endoplasmic reticulum (ER) and secrete outside cell membrane then form to extracellular oligometric matrix proteins and collagens. All is known that the ER is involved in a variety of important biological activities, including protein secretion, lipid synthesis, and calcium regulation [1,2]. As a result, an efficient regulation of ER activity is important to the survival of the organism in response to changes in the physiological environment. Conditions that disrupt ER function can decrease the ER's protein folding ability, resulting in the accumulation and aggregation of unfolded protein [3-6]. Cells engage a biochemical program termed an Unfolded Protein Response (UPR) route to counteract the detrimental consequences of ER stress. The UPR is involved in regulating the process of differentiation and growth of osteoblast and chondrocyte within the skeletal system [7-9]. The ER of osteoblast and chondrocyte has a well-established mechanism, which can improve their ability for UPR protein production during ER stress response.
The UPR comprises three signaling pathways triggered by three distinct transmembrane sensors– inositol-requiring enzyme 1 (IRE1,encoded by ERN1), activating transcription factor 6 (ATF6), and protein kinase R–like endoplasmic reticulum kinase (PERK, encoded by EIF2AK3) [10-12]. IRE1α is the UPR evolution's most conservative branch [13]. After the activation of IRE1α under ER stress, it clues specific exon intron sites into an mRNA, encrypting the transcription factor X-box-batches protein 1 (XBP1). This enhances the protein-folding capacity of the endoplasmic reticule and reduces the misfolded pro-ER1. This results in the production of active transcription factors and induction of different adaptive genes [14]. At the same time, the active IRE1α may also decrease the quantity of protein entering an ER via particular ER-localized cytoplasmic mRNA. This mechanism is known as IRE1-dependent decay control (RIDD)[13]. The JNK, ERK, and NF-κB path can also be activated by IeR1α through interaction with several adapters and regulatory proteins [15-17]. ATF6 is an endoplasmic transmembrane reticulum protein having in its cytoplasmic field bZIP transcription factor [18]. ATF6 translates under ER pressure into the Golgi apparatus, where it is split into two sites with the proteases site 1 (S1P) and site cleavage-2 (S2P). The ATF6 (ATF6-N) released amino terminus migrates to the nucleus and activates the chaperons for ER encoding, including those genes that increase the ERAD pathway. ATF6 can also be used to promote gene-expression patterns between Heterodimers and XBP1 [19]. PERK is a serine/threonine kinase that oligomerizes and trans-autophosphorylates under ER stress conditions, inhibiting general protein translation via phosphorylation of eukaryotic translation initiator factor-2 (eIF2) at serine, while selectively increasing ATF4 translation, and then reducing protein load into the ER [19]. ATF4 then activates a set of genes involved in regulating of antioxidant responses, ER folding capacity, amino acid metabolism, antioxidant responses, and macro-autophagy [13,18].
Obviously, eukaryotic cells have the above-mentioned signaling pathways from the ER to entire cell, to avoid overabundant cumulation of misfolded and unfolded proteins in the ER and maintain the ER homeostasis. Based on the literature, we assessed the bone metabolism-related proteins involved in the three pathways of UPR, and the role of these UPR signaling molecules in regulating bone metabolism biomarkers, providing a basis for the subsequent development of biologics and diagnosis and treatment.
Overview of Molecular Markers of IRE1Α-XBP1 Pathway Involved in Bone Metabolism (Table 1)
Overexpression of IRE1α suppresses the differentiation of collagen II (ColII), SOX9, collagen X (ColX), metalloproteinase matrix 13 (MMP-13), hedgehog indian (IHH), RUNX2, and increased parathyroid-related peptide expression (PTHrP) [20]. The deficiency of IRE1α in chondrocytes downregulates prosurvival factors XBP1S and Bcl-2, which enhances the apoptosis of chondrocytes through increasing proapoptotic factors caspase-3, p-JNK, and CHOP. Meanwhile, the activation of IRE1α increases chondrocyte viability and reduces cell apoptosis [21]. The in vitro expression of cartilage formation indicators such as ColII, ColX and RUNX2, SOX9, and XBP1S can promote efficient cartilage development. Furthermore, XBP1S can also activate the precursor of granulin-epithelin (GEP, which increases the development of cartilages), which favorably regulates chondrocyte hypertrophy, mineralization and bone formation [22,23]. Besides, Chondrocyte apoptosis and proliferation are affected by the deletion of XBP1 in mice, and XBP1 signaling pathway is essential for alleviating mutant protein aggregation in ER-stress related skeletal disease, it presents a therapeutic target for aggregation related conditions in cells undergoing proliferation [24]. A high-throughput sequencing result found that there are multiple ncRNAs in cartilage XBP1 knockout mice, and these differentially expressed ncRNAs help to clarify the occurrence and development of bone development-related diseases and help identify prospective clinical markers [25].
|
References |
Published Date/ Journal |
Authors |
Targets |
|
The IRE1α-XBP1 pathway is essential for osteoblast differentiation through promoting transcription of Osterix |
2011 Mar/ EMBO Rep. |
Tohmonda T, Miyauchi Y, Ghosh R, et al. |
IRE1, XBP1, Osterix, Gadd34, CHOP/Gadd153, Ero1a, Wfs1 |
|
Unfolded protein response mediator, the IRE1α-XBP1 pathway is involved in osteoblast differentiation. |
|
Tohmonda T, Chiba K, Toyama Y, et al. |
IRE1α-XBP1, Osterix |
|
The IRE1α-XBP1 pathway positively regulates parathyroid hormone (PTH)/PTH-related peptide receptor expression and is involved in pth-induced osteoclastogenesis. |
2013 Jan/ J Biol Chem. |
Tohmonda T, Yoda M, Mizuochi H, et al. |
parathyroid hormone (PTH)/PTH-related peptide receptor (PTH1R), IRE1α-XBP1, RANK1 |
|
Toll-like receptor-mediated IRE1α activation as a therapeutic target for inflammatory arthritis. |
2013 Aug/ EMBO J. |
Qiu Q, Zheng Z, Chang L, et al. |
IRE1α, tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6), protein phosphatase 2A (PP2A) |
|
XBP1S, a BMP2-inducible transcription factor, accelerates endochondral bone growth by activating GEP growth factor |
2014 Mar/ J Cell Mol Med. |
Guo FJ, Xiong Z, Han X, et al |
GEP, RUNX2 |
|
Cartilage-specific ablation of XBP1 signaling in mouse results in a chondrodysplasia characterized by reduced chondrocyte proliferation and delayed cartilage maturation and mineralization |
2015 Apr/ Osteoarthritis Cartilage. |
Cameron TL, Gresshoff IL, Bell KM, et al. |
collagen II, collagen X, IRE1 |
|
IRE1α/XBP1-mediated branch of the unfolded protein response regulates osteoclastogenesis. |
2015 Jul/ J Clin Invest. |
Tohmonda T, Yoda M, Iwawaki T, et al. |
NFATc1, ITPR2 and ITPR3, XBP1 |
|
XBP1-Independent UPR Pathways Suppress C/EBP-β Mediated Chondrocyte Differentiation in ER-Stress Related Skeletal Disease |
2015 Sep/ PLoS Genet. |
Cameron TL, Bell KM, Gresshoff IL, et al. |
IRE1/XBP1, CEBPB |
|
Chondroprotective effects of alpha-lipoic acid in a rat model of osteoarthritis |
2016 May/ Free Radic Res. |
Wang J, Sun H, Fu Z, et al. |
caspase proteins, NADPH oxidase 4 (Nox4), p22(phox), activation of nuclear factor-κB (NF-κB) |
|
Endoplasmic reticulum stress cooperates with Toll-like receptor ligation in driving activation of rheumatoid arthritis fibroblast-like synoviocytes |
2017 Sep/ Arthritis Res Ther. |
Kabala PA, Angiolilli C, Yeremenko N, et al. |
recognizing histone 3, IκBα, and phosphorylated forms of JNK, p38, ERK, CHOP, BiP, TLR signaling |
|
ER Stress: A Therapeutic Target in Rheumatoid Arthritis? |
2018 Apr/ Trends Pharmacol Sci. . |
Rahmati M, Moosavi MA, McDermott MF. |
GRP78/BiP, IRE1α, PERK, ATF6, ERAD |
|
Endoplasmic Reticulum Stress Signalling During Development |
2019 Mar/ In: Clarke R, ed. |
Dominicus CS, Patel V, Chambers JE, et al. |
Unfolded Protein Response, vascular endothelial growth factor A (VEGF-A) |
|
XBP1 signaling is essential for alleviating mutant protein aggregation in ER-stress related skeletal disease. |
2019 Jul/ PLoS Genet. |
Piróg KA, Dennis EP, Hartley CL, et al. |
IRE1, ATF6, PERK, Pdia6, Creld2, Derl1, GRP94, MANF, DDIT3 |
|
The Increased RNase Activity of IRE1α in PBMCs from Patients with Rheumatoid Arthritis. |
2019 Aug/ Adv Pharm Bull. |
Ahmadiany M, Alavi-Samani M, Hashemi Z, et al. |
GRP78, IRE1, XBP1S, regulated IRE1-dependent decay (RIDD) targets (miRNA-17, -34a, -96, and -125b) |
|
New developments in chondrocyte ER stress and related diseases. |
2020 Apr/ F1000Res. |
Briggs MD, Dennis EP, Dietmar HF, et al. |
PREK, IRE1- α, ATF6α, BiP, CHOP, caspase-12 |
|
ER stress arm XBP1S plays a pivotal role in proteasome inhibition-induced bone formation. |
2020 Nov/ Stem Cell Res Ther. |
Zhang D, De Veirman K, Fan R, et al. |
IRE1α-XBP1S, PERK-ATF4, Col1a1, Ocn, Bmp2, RUNX2, Opn, Grp78, CHOP |
|
IRE1 signaling regulates chondrocyte apoptosis and death fate in the osteoarthritis. |
2021 Jul/ J Cell Physiol. |
Huang R, Hui Z, Wei S, et al |
caspase-3, p-JNK, CHOP |
|
The Role of Unfolded Protein Response in Human Intervertebral Disc Degeneration: Perk and IRE1- α as Two Potential Therapeutic Targets |
2021 Mar/ Oxid Med Cell Longev. |
Wen T, Xue P, Ying J, et al. |
PREK, IRE1- α, ATF6, GRP78, CHOP |
|
IRE1-mTOR-PERK Axis Coordinates Autophagy and ER Stress-Apoptosis Induced by P2X7-Mediated Ca(2+) Influx in Osteoarthritis. |
2021 Jun/ Front Cell Dev Biol. |
Li Z, Huang Z, Zhang H, et al |
PREK, IRE1α, ATF6, GRP78, CHOP, mTOR, p-PERK, p-IRE1, LC3B, Beclin-1, LAMP2, caspase-12 |
|
Effect Of XBP1 Deficiency In Cartilage On The Regulatory Network Of LncRNA/circRNA-miRNA-mRNA |
2022 Jan 1/Int J Biol Sci. |
Xiaoli Li, Yuyou Yang, et al |
XBP1S, LncRNA/circRNA-miRNA-mRNA |
OA is a degenerative common ailment that has an important impact on the quality of life of aged people, causing annoyance, pain, and sometimes disability [26]. The breakdown of cartilage in OA is characterized by aberrant cartilage cell metabolism and extracellular matrix degradation [27]. Chondrocyte metabolic balance and OA symptoms can be influenced by altering ER stress and autophagy by mechanical loading [28]. In the lumen of ER, excessive mechanical stress induces Ca2+ overflow, accumulation of malfunctioning proteins in the ER lumen and ER stress induction [28]. In addition, molecular chaperone disorders can also cause severe ER stress, which will lead to severe forms of OA, accompanied by increased cell death of chondrocytes [29]. ER stress was observed in chondrocytes in osteoarthritis (OA) patients, and ER stress and apoptosis increased throughout OA development [30]. In moderate OA cartilage, but not in mild or severe cartilage, XBP1 mRNA splicing has been enhanced [31]. Researchers also thought that the signal axis of IRE1-mTOR-PERK is used to control OA-derived inhibition and activation of Apoptosis [32]. The IRE1 driven autophagy flow is stopped by mTOR, and PERK blocks the subsequent fusion of automotive phagosomes and lysosomes. mTOR promotes PERK to enhance caspase-12 and CHOP production and to induce apoptosis of ER-stressed cells. Chain protein (BiP) expression has dramatically enhanced following medial meniscus (DMM) surgery in wild-type mouses and shows clearly that ER stress and UPR are implicated in OA development [33]. After DMM surgery in wild-type mice, BiP expression increased significantly, clearly indicating that ER stress and UPR are involved in the pathogenesis of OA. Although, a misfolded version of thyroglobulin (Tg cog) was overexpressed under the Col2a1 promoter (ColIITg cog, a model of cartilage-specific ER stress [34]) in transgenic mice following the DMM operation, BiP levels have also been increased but OA ontake have been delayed, and a delay in chondrocyte apoptosis and damage two weeks after surgical operation were also found in comparison with wild-type control. Before DMM surgery, ColIITg cog mice increased its immunoglobulin heavy BiP level and the ColIITg cog mice pro-survival XBP1 network of signals was activated in ColIITg cog mice after DMM [34], indicating that the ER stress pre-exposure or a specific pro-survival UPR response was activated. Chondroprotective impact may occur in the XBP1S signal. Besides, XBP1S inhibits ER stress-mediated apoptosis and nitrite production in OA cartilage, and XBP1S also acts as a negative regulator of apoptosis in osteoarthritis by affecting caspase 3, caspase 9, caspase 12, p-JNK1, and the pro-apoptotic ER stress marker C/EBP homologous protein (CHOP) [35]. Based on detailed analysis of gene expression patterns some researchers proposed that XBP1-independent UPR driven disruption of CCAAT/enhancer-binding protein beta (CEBPβ or CEBPB, a master regulator of chondrocyte differentiation) is important for the skeletal disease pathophysiology [36].
We sent human chondrocytes overexpressing IRE1α to Kangcheng Biotech for differential expression profiling chip (DEPC) and ChIP-chip transcription factor microarray (CTFM), Raw signal intensities were normalized in the RMA method by NimbleScan v2.5, and low-intensity genes were filtered (Genes with Intensity ≥ 100.0 in at least 2 out of 3 samples were chosen for further analysis). Differentially expressed genes that passed Fold Change filtering (Fold Change ≥ 2.0). The DEPC results uncovered that after overexpressing IRE1a, the expression profile of 532 genes changed, 19 of which are related to bone development or bone-related diseases (Figures 1A and 1B). Then, Gene Ontology (GO), kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis was performed on these 19 differential genes. These genes are mainly involved in the following four aspects (Figure 2A): Osteogenesis imperfecta, bone trabecula formation, bone marrow development, and negative regulation of chondrocyte differentiation, and avascular necrosis of femoral head. Furthermore, the CTFM results show that XBP1S can directly regulate the development of bone through the pathway of ossification, osteoclast fusion, proliferation, differentiation etc. (Figure 3A). In these signal pathways, XBP1S can bind to the growth factor of connective tissue (CTGF) to control chondrocyte differentiation and proliferation or in combination with Oxytocin (OXT), to regulate ossification involved in bone maturation by bind to ras homolog gene family member A (RHOA) or in combination with TC cell leukemia (TCTA) to regulate osteoclast fusion (Figure 4).
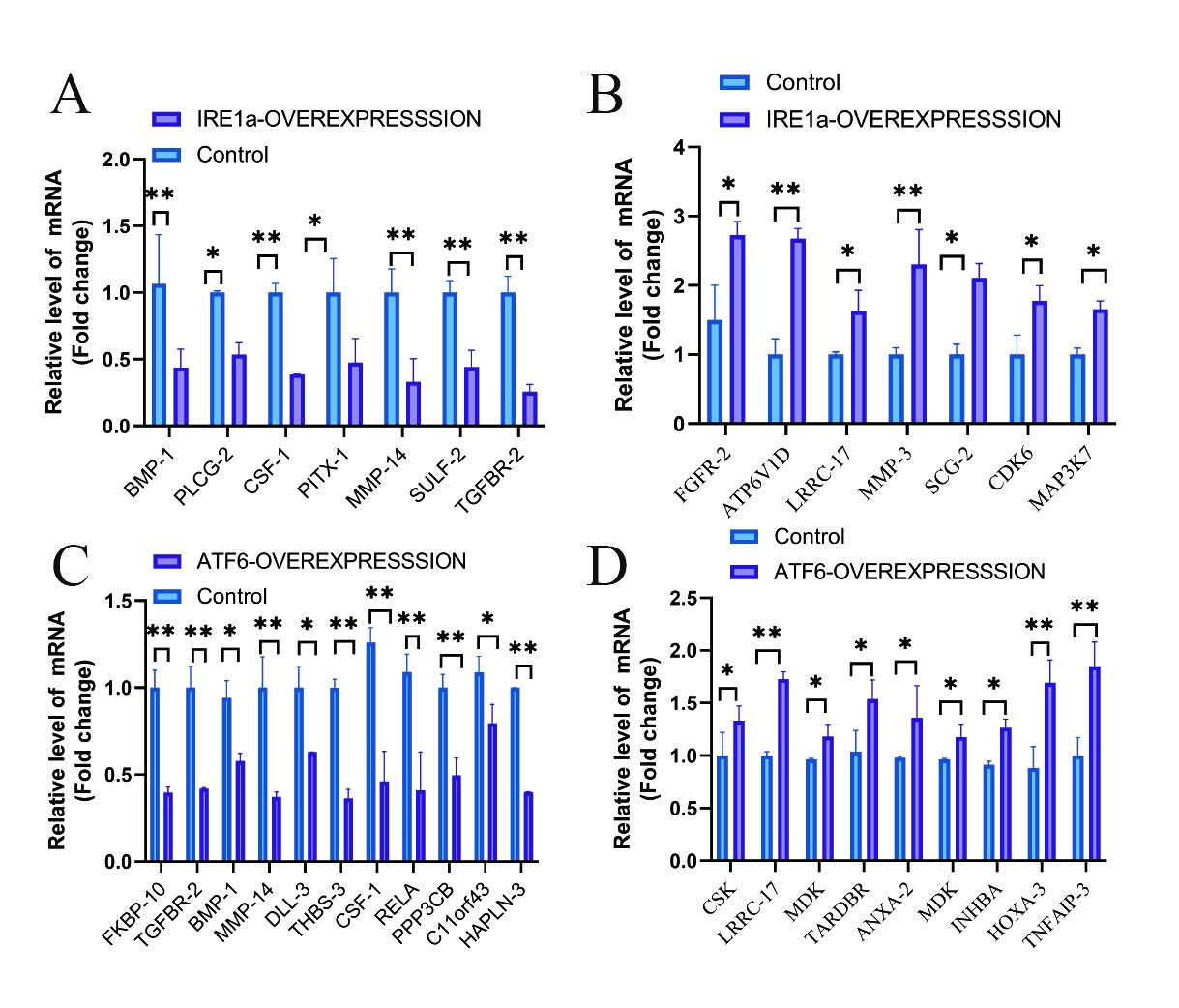
Figure 1: The differential expression profiling analysis of mRNA levels of bone development and bone-related diseases genes affected by different UPR molecules. A, B: Quantitative analysis of mRNA levels after IRE1α overexpression in human chondrocytes. C, D: Quantitative analysis of mRNA levels after ATF6 overexpression in human chondrocytes. The error bars represent the SEM. *p < 0.05, **p < 0.01.
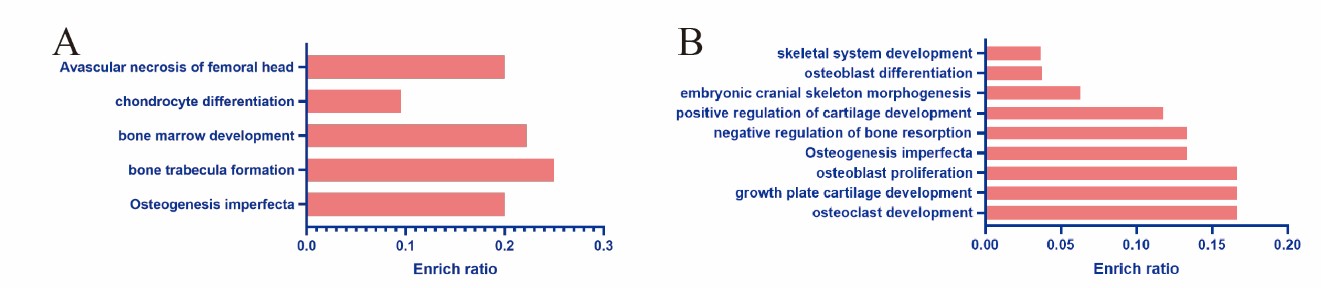
Figure 2: Enriched terms visualized in bar-plot of differential expression profiling chip. Each row represents an enriched function, and the length of the bar represents the enrich ratio, which is calculated as “input gene number”/ “background gene number”. A: Enrichment of gene function regulated by overexpression IRE1α in human chondrocytes, B: Enrichment of gene function regulated by overexpression ATF6 in human chondrocytes.
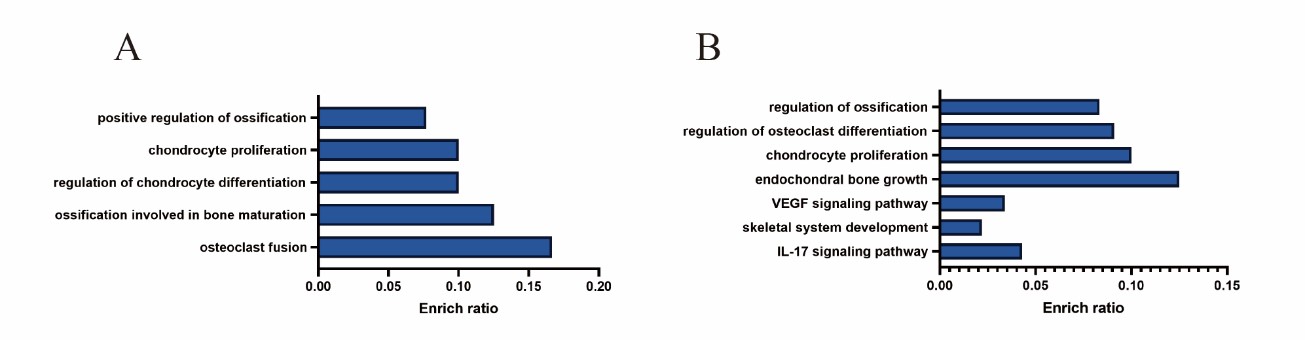
Figure 3: Enriched terms visualized in barplot of ChIP-chip transcription factor Microarray. Each row represents an enriched function, and the length of the bar represents the enrich ratio, which is calculated as “input gene number”/ “background gene number”. A: Enrichment of gene function regulated by transcription factor XBP1S binding, B: Enrichment of gene function regulated by transcription factor ATF6 binding.
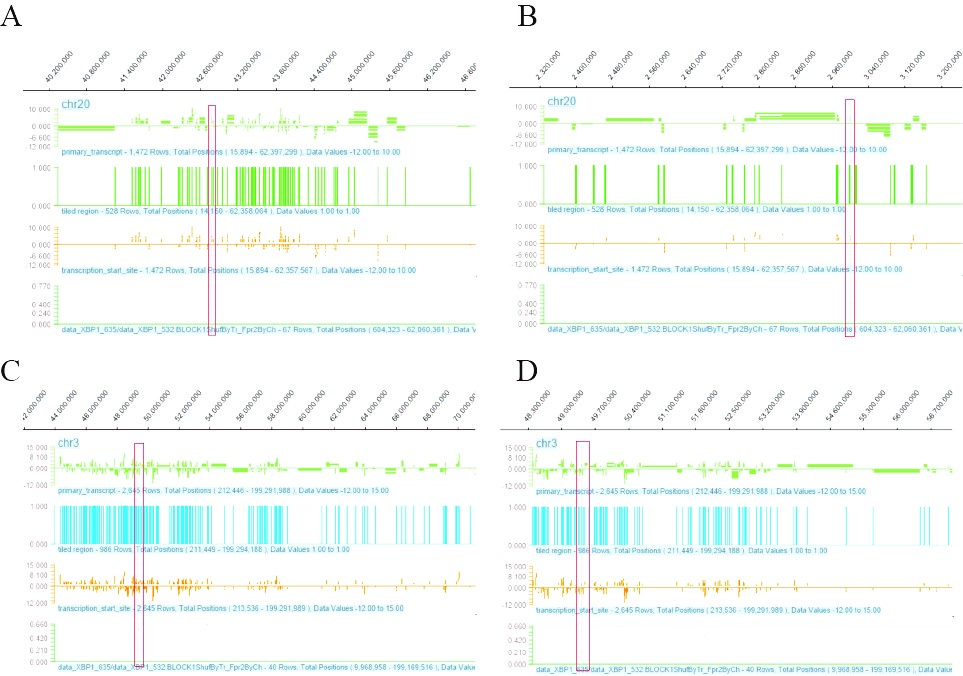
Figure 4: The ChIP-chip Microarray analysis of the promoter region of the transcription factor XBP1S binding gene in chondrocytes. A:XBP1S interacts with the promoter region of CTGF gene, the red box shows the binding site; B: XBP1S interacts with the promoter region of OXT gene, the red box shows the binding site; C: XBP1S interacts with the promoter region of RHOA gene, the red box shows the binding site; D: XBP1S interacts with the promoter region of TCTA gene, the red box shows the binding site.
XBP1 is critical for bone morphogenetic protein 2 (BMP2) induced osteoblast differentiation. If without IRE1, BMP2 therapy failed to augment the activity of alkaline phosphatase or expression of the osteoblast markers Osterix (a bone forming transcription factor required) and osteocalcin [8].The IRE1α-XBP1 pathway can activate Osterix transcript, whereas XBP1 binds directly to the promoter region of the Osterix gene [37]. Signaling XBP1 inhibition may severely damage osteogenic differentiation caused by proteasomic inhibitors (PIs). In addition, XBP1S able to transcript the expression of ATF4, while XBP1S' overexpression can induce the expression of ATF4 and other osteogenic differentiation genes including Col1a1, Osteocalcin (Ocn), Bone morphogenetic protein 2 (Bmp2, RUN2), and Osteopontin (Opn), thus driving different types of osteoblasts. The results show that XBP1 may be late in the process of differentiation of the osteoblast [38].
The removal of IRE1 from bone marrow cells caused an increase in bone mass was unexpected [39]. This is because XBP1 contacts the promoter directly and promotes gene transcription that encodes the nuclear coding of cytoplasmic 1 activated t-cells (NFATc1), a master regulator for bone absorbing osteoclasts [39]. Therefore, IRE1α deletion is linked to the poor differentiation of the osteoclastic. The xbp1 mRNA processing is considerably decreased by pharmacological inhibition or detection of these receptors and may be transiently activated during osteoclastogenesis, partially dependent upon Ca2+ oscillation mediated by inositol 1,4,5-trisphosphate receptors 2 and 3, (ITPR2 and ITPR3) in the reticulum of the endoplasma. The NF-κB ligand (RANK1) gene encodes an osteoclast factor secreted by osteoblasts, and the receptor activator of Rank1 can be induced to express by PTH signal [40]. BMP2-mediated Pth1r transcription increase in mouse embryonic fibroblasts can reverse silences of the Ire1α and XBP1 genes, as XBP1 is capability to adjust Pth1r transcription directly. In addition, PTH-induced Rank1 expression in primary osteoblasts can be suppressed by silencing of XBP1 gene, thus osteoclastic development in an in vitro osteoclastogenesis model is eliminated [41].
ATF6 Pathway Participates in the Regulation of Molecular Markers of Bone Metabolism (Table 2)
Enhanced chondrocyte hypertrophy, mineralization, and endochondral bone by overexpression of ATF6 in mice [42], and knock-down of ATF6 prevents differentiation of chondrocytes hypertrophy [43]. BMP2 enhances the differentiation of osteoblast and mineralization by expressing ATF6 triggered by RUNX2, which in turn promotes osteocalcin transcription [44]. ATF6α also improves the chondrocyte hypertrophy of RUNX2 and changes the pathway to in vitro production of the crucial Indian hedgehog and parathyroid hormone cartilage [43]. Interestingly, osteocalcin expression has risen following co-expression of ATF6, RUNX2, and ATF4, indicate that ATF6 may have a synergistic impact on expression of osteocalcin [45]. Furthermore, ATF6 overexpression in human dental pulp cells shows that distinction between dentistry and matrix mineralization may be regulated by it [46]. ATF6 aggravates angiogenesis-osteogenesis coupling during ankylosing spondylitis by mediating fibroblast growth factor 2 (FGF2) expression in chondrocytes [47]. The SLC26A2 gene is responsible for a variety of presently incurable human chondrodysplasias, including sarcomas. In SLC26A2-deficient chondrodysplasias, suppressing ATF6-dependent overactivation of the fibroblast growth factor receptor 3 (FGFR3) signaling ameliorates the condition [48].
| References |
Published Date/ Journal |
Authors | Targes |
|---|---|---|---|
|
Suppressing UPR-dependent overactivation of FGFR3 signaling ameliorates SLC26A2-deficient chondrodysplasias |
2019 Feb/ EBioMedicine. |
Zheng C, Lin X, Xu X, et al. |
ATF6, FGFR3, p-ERK1/2, ERK1/2 |
|
Celastrol ameliorates endoplasmic stress-mediated apoptosis of osteoarthritis via regulating ATF-6/CHOP signaling pathway |
2020 Jun/ J Pharm Pharmacol. |
Liu DD, Zhang BL, Yang JB, et al. |
ATF-6/CHOP signaling pathway |
|
|
|
|
|
|
ATF6 aggravates angiogenesis-osteogenesis coupling during ankylosing spondylitis by mediating FGF2 expression in chondrocytes |
2021 Jun/ iScience. |
Ma M, Li H, Wang P, et al. |
tumor necrosis factor alpha (TNF-α), interferon-γ (IFN-γ) or interleukin-17 (IL-17), FGF2 |
|
BMP2 protein regulates osteocalcin expression via RUNX2-mediated Atf6 gene transcription. |
2011 Nov/ J Biol Chem. |
Jang WG, Kim EJ, Kim DK, et al. |
RUNX2, ATF6, Oasis |
|
TNFα modulates protein degradation pathways in rheumatoid arthritis synovial fibroblasts. Arthritis |
2012 Mar/ Arthritis Res Ther. |
Connor AM, Mahomed N, Gandhi R, et al. |
GRP78/BiP |
|
ATF6 upregulates XBP1S and inhibits ER stress-mediated apoptosis in osteoarthritis cartilage |
2013 Nov/ Cell Signal. |
Guo FJ, Xiong Z, Lu X, et al. |
caspase 3, caspase 9, caspase 12, p-JNK1, and CHOP |
|
Transcriptional factor ATF6 is involved in odontoblastic differentiation. |
2014 May/ J Dent Res. |
Kim JW, Choi H, Jeong BC, et al. |
ATF6, dentin sialophosphoprotein (DSPP) and dentin matrix protein 1 (DMP1), |
|
ATF6a, a RUNX2-activable transcription factor, is a new regulator of chondrocyte hypertrophy. |
2016 Feb/ J Cell Sci. |
Guo F, Han X, Wu Z, et al. |
RUNX2, PTHLH signaling, Sox6 |
|
Atf6α deficiency suppresses microglial activation and ameliorates pathology of experimental autoimmune encephalomyelitis. |
2016 Dec/ J Neurochem. |
Ta HM, Le TM, Ishii H, et al. |
ATF6α, glucose-regulated protein 78 (GRP78), NF-κB p65 |
|
Paradoxical roles of ATF6α and ATF6β in modulating disease severity caused by mutations in collagen X. |
2018 Mar/ Matrix Biol. |
Forouhan M, Mori K, Boot-Handford RP. |
collagen X, ATF6 |
|
A lifetime of stress: ATF6 in development and homeostasis. |
2018 May/ J Biomed Sci. |
Hillary RF, FitzGerald U. |
ATF6 axis |
|
A novel pathogenic role of the ER chaperone GRP78/BiP in rheumatoid arthritis. |
2019 Aug/ J Exp Med. |
Yoo SA, You S, Yoon HJ, et al. |
MMP-13, Circ0136474, miR-127-5p |
|
Cartilage endoplasmic reticulum stress may influence the onset but not the progression of experimental osteoarthritis. |
2019 Sep/ Arthritis Res Ther. |
Kung LHW, Mullan L, Soul J, et al. |
ATF6α, Col2a1, BiP |
The ATF6α route in OA, however, appears not crucial. Although the ER stress induction and the signaling of XBP1 has been demonstrated to be associated to ATF6α [35,42], ATF6α knockout mice may not be as relevant as other UPR branches, having normal articular cartilage and developing the same rates as wild types of controls after DMM [33]. The ADAMTS-5 and ADAMTS-4 expression, nevertheless, was reduced by a silencing of ATF6β [49]. Celastrol ameliorates endoplasmic stress-mediated apoptosis of OA via regulating ATF-6/CHOP signaling pathway [50], thus demonstrating how important a greater knowledge of the OA development is to the UPR.
Similarily, we also sent human chondrocytes overexpressing ATF6 to Kangcheng Biotech for DEPC and CTFM. The DEPC results showed that the expression of 362 genes has changed, 23 of which are related to bone development or bone-related diseases (Figures 1C and 1D). Analysis of these 23 differential genes using GO and KEGG pathways revealed that these genes are mostly engaged in the following aspects (Figure 2B): osteoclast development, growth plate cartilage development, osteoblast proliferation, osteogenesis imperfecta, negative regulation of bone resorption, positive regulation of cartilage development, embryonic cranial skeleton morphogenesis, osteoblast differentiation, skeletal system development, and Osteoclast differentiation. Besides, the ATF6 CTFM results found that ATF6 can directly regulate chondrogenesis and bone development through the pathway of endochondral bone growth, proliferation, differentiation and ossification, including vascular endothelial growth factor (VEGF) and IL17 signaling pathway (Figure 3B). ATF6 can directly bind to frizzled-related protein (FRZB), ubiquitin specific peptidase 1(USP1), FGFR3, CEBPB, and mitogen-activated protein kinase 1 (MAPK1) to directly regulate skeletal system development, endochondral bone growth, chondrocyte proliferation, and regulation of osteoclast differentiation and ossification. In these pathways, ATF6 can directly bind to CEBPB, MAPK1, splicing factor, arginine/serine-rich 1(SFRS1) or mitogen-activated protein kinase 13(MAPK13) in the IL-17 signaling pathway to directly regulate it (Figure 5).
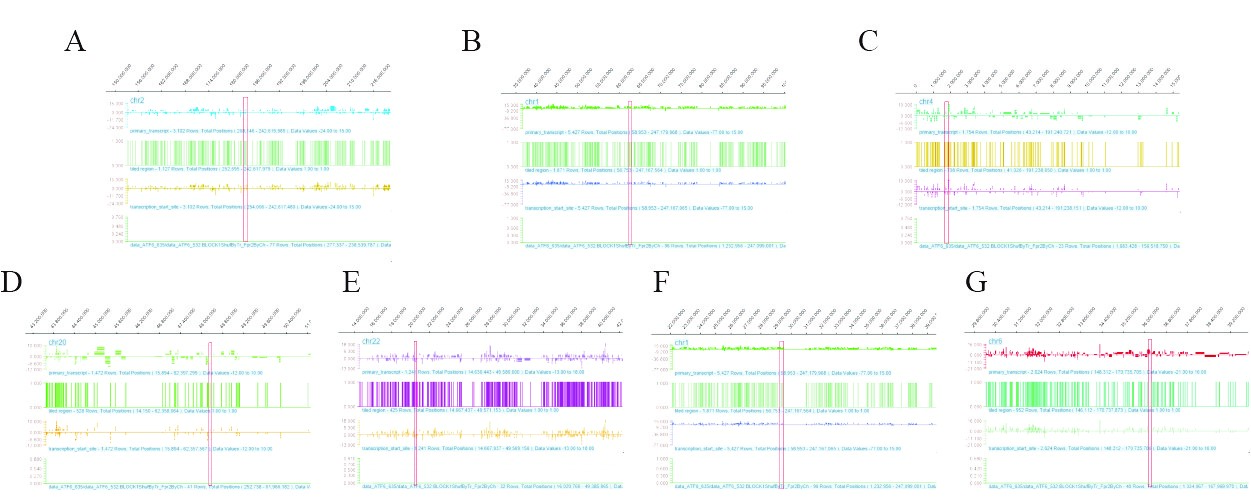
Figure 5: The ChIP-chip Microarray analysis of the promoter region of the transcription factor ATF6 binding gene in chondrocytes. A: ATF6 interacts with the promoter region of FRZB gene, the red box shows the binding site; B: ATF6 interacts with the promoter region of USP1 gene, the red box shows the binding site; C: ATF6 interacts with the promoter region of FGFR3 gene, the red box shows the binding site. D: ATF6 interacts with the promoter region of CEBPB gene, the red box shows the binding site; E: ATF6 interacts with the promoter region of MAPK1 gene, the red box shows the binding site; F: ATF6 interacts with the promoter region of SFRS1 gene, the red box shows the binding site; G: ATF6 interacts with the promoter region of MAPK13 gene, the red box shows the binding site.
Overview of Molecular Markers of UPR Pathway Involved in Bone Metabolism (Table 3)
In OA samples the ERK (PERK)1/2-positive phosphorylated chondrocytes were considerably greater than in healthy controls which suggested the pathological role of that pathway in OA [51]. The breakdown in the PERK pathway in OA chondrocytes with the accompanying COL2A1 expression reduction was identified leading to the extracellular matrix disintegration. The reduction in PERK in OA chondrocytes causes collagen II to be reduced and collagen I to be enhanced [29]. Loss of collagen II and increased collagen I expression are known to interfere with articular cartilage function [34]. Besides, Diazoxide prevents chondrocyte apoptosis and cartilage degeneration OA by PERK1/2 and ERK1/2 signaling pathways [52]. Sirtuin-1 (SIRT1), a NAD+-dependent deacetylase, is dysregulated in osteoarthritis. SIRT1 is a highly essential regulator of cartilage production and maintenance of cartilage homeostasis, it deacetylates PERK and weakens the PERK-eIF-2-CHOP-axis to enhance cartilage production and bone growth [53]. Under these results, weakened PERK can be an OA biomarker and increasing PERK can therapeutically develop OA [54]. Studies by Botter., Glasson. et al. have shown that Aggrecanase 2 deficient (ADAMTS5-/-) mice can be protected against OA-caused cartilage injury [55]. After overexpression of IREla, we found that the expression of ADAMTS5 decreased significantly. This also provides support for UPR signaling molecules as a biological indicator of OA.
|
References |
Published Date/ Journal |
Authors |
Targes |
|
Endoplasmic reticulum stress response mediated by the PERK-eIF2(alpha)-ATF4 pathway is involved in osteoblast differentiation induced by BMP2 |
2011 Feb/ J Biol Chem. |
Saito A, Ochiai K, Kondo S, et al. |
PERK-eIF2(alpha)-ATF4 pathway |
|
BMP2 protein regulates osteocalcin expression via RUNX2-mediated Atf6 gene transcription. |
2012 Jan/ J Biol Chem. |
Jang WG, Kim EJ, Kim DK, et al. |
RUNX2, ATF6, osteocalcin |
|
A functional haplotype in EIF2AK3, an ER stress sensor, is associated with lower bone mineral density. |
2012 Feb/ J Bone Miner Res. |
Liu J, Hoppman N, O'Connell JR, et al. |
EIF2AK3, ER stress sensor |
|
A novel pathogenic role of the ER chaperone GRP78/BiP in rheumatoid arthritis. |
2012 Apr/ J Exp Med. |
Yoo SA, You S, Yoon HJ, et al. |
GRP78/BiP |
|
Osteoporosis regulation by salubrinal through eIF2α mediated differentiation of osteoclast and osteoblast. |
2013 Feb/ Cell Signal. |
He L, Lee J, Jang JH, et al. |
NFATc1, RANKL, eIF2α, ATF4 |
|
Combination of MEK-ERK inhibitor and hyaluronic acid has a synergistic effect on anti-hypertrophic and pro-chondrogenic activities in osteoarthritis treatment. |
2013 Mar/ J Mol Med (Berl). |
Prasadam I, Mao X, Shi W, et al. |
collagen X, RUNX2, ADAMTs5, MMP-13 |
|
Hyperglycemia induces endoplasmic reticulum stress-dependent CHOP expression in osteoblasts |
2013 May/ Exp Ther Med. |
Liu W, Zhu X, Wang Q, et al. |
CHOP |
|
ATF4 promotes bone angiogenesis by increasing VEGF expression and release in the bone environment. |
2013 Sep/ J Bone Miner Res. |
Zhu K, Jiao H, Li S, et al. |
ATF4, hypoxia-inducible factor 1α (HIF-1α), vascular endothelial growth factor (VEGF) |
|
VEGF Signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress. |
2014 May/Mol Cell. |
Karali E, Bellou S, Stellas D, et al. |
ATF6, PERK, AKT, CHOP, eIF2α |
|
17β-Estradiol inhibits ER stress-induced apoptosis through promotion of TFII-I-dependent Grp78 induction in osteoblasts |
2014 Jun/ Lab Invest. |
Guo YS, Sun Z, Ma J, et al. |
17β-E2, Grp78, caspase-12 , caspase-3, Ras-ERK1/2-TFII-I signaling pathway |
|
Bone tissue remodeling and development: Focus on matrix metalloproteinase functions. |
2014 Nov/ Arch Biochem Biophys. |
Paiva KBS, Granjeiro JM. |
Matrix metalloproteinases (MMPs) |
|
Novel mechanism of enhancing IRE1α-XBP1 signaling via the PERK-ATF4 pathway. |
2016 Apr/Sci Rep. |
Tsuru A, Imai Y, Saito M, et al. |
IRE1α-XBP1 signaling, PERK-ATF4 pathway |
|
Role of Endoplasmic Reticulum Stress and Unfolded Protein Responses in Health and Diseases. |
2016 Apr/ Indian J Clin Biochem. |
Mahdi AA, Rizvi SH, Parveen A. |
Endoplasmic reticulum (ER), unfolded protein responses (UPR) |
|
The unfolded protein response genes in human osteoarthritic chondrocytes: PERK emerges as a potential therapeutic target. |
2016 Jul/Arthritis Res Ther. |
Li YH, Tardif G, Hum D, et al. |
ERN1, PERK, CREB3L2, COL2a1, MMP-13, ADAMTS4 and ADAMTS5 |
|
The unfolded protein response in skeletal development and homeostasis. |
2016 Aug/ Cell Mol Life Sci. |
Horiuchi K, Tohmonda T, Morioka H. |
UPR |
|
Role of endoplasmic reticulum stress in disuse osteoporosis. |
2017 Apr/ Bone. |
Li J, Yang S, Li X, et al. |
Bip, p-eIF2α, CHOP and ATF4 |
|
Cartilage-Specific Autophagy Deficiency Promotes ER Stress and Impairs Chondrogenesis in PERK-ATF4-CHOP-Dependent Manner |
2017 Oct /J Bone Miner Res. |
Kang X, Yang W, Feng D, et al. |
PERK-ATF4-CHOP axis, ATG7, col10a1, MMP13, VEGFA |
|
Diazoxide prevents H 2 O 2-induced chondrocyte apoptosis and cartilage degeneration in a rat model of osteoarthritis by reducing endoplasmic reticulum stress |
2017 Nov/ Biomed Pharmacother. |
Gu Y, Chen J, Meng Z, et al. |
PERK1/2 and ERK1/2 signaling pathways, tcaspase-3, Bax, ATF-6, CHOP, Bcl-2, collagen II |
|
Sirtuin-1 (SIRT1) stimulates growth-plate chondrogenesis by attenuating the PERK-eIF-2α-CHOP pathway in the unfolded protein response |
2018 Jun /J Biol Chem. |
Kang X, Yang W, Wang R, et al. |
PERK-eIF-2α-CHOP axis, SIRT1 |
|
Defective autophagy in osteoblasts induces endoplasmic reticulum stress and causes remarkable bone loss |
2018 Jul/ Autophagy |
Li H, Li D, Ma Z, et al. |
TNFSF11, RANKL, TNFRSF11B, OPG, ATG7, DDIT3, CHOP, MAPK8, JNK1, SMAD1/5/8 |
|
Expression of UPR effector proteins ATF6 and XBP1 reduce colorectal cancer cell proliferation and stemness by activating PERK signaling. |
2019 Jun/ Cell Death Dis. |
Spaan CN, Smit WL, van Lidth de Jeude JF, et al. |
ATF6, XBP1, PERK |
|
Curcumin/Liposome Nanotechnology as Delivery Platform for Anti-inflammatory Activities via NFkB/ERK/pERK Pathway in Human Dental Pulp Treated With 2-HydroxyEthyl MethAcrylate (HEMA). |
2019 Jun/ Front Physiol. |
Sinjari B, Pizzicannella J, D'Aurora M, et al. |
NFkB/ERK and pERK signaling |
|
Role of endoplasmic reticulum stress in rheumatoid arthritis pathogenesis. |
2019 Sep/ J Korean Med Sci. |
Park YJ, Yoo SA, Kim WU. |
ER stress, GRP78 |
|
4-Phenylbutyric Acid Reduces Endoplasmic Reticulum Stress in Chondrocytes That Is Caused by Loss of the Protein Disulfide Isomerase ERp57. |
2019 Oct/Oxid Med Cell Longev. |
Rellmann Y, Gronau I, Hansen U, et al. |
ER stress |
|
Knockdown of cytokeratin 8 overcomes chemoresistance of chordoma cells by aggravating endoplasmic reticulum stress through PERK/eIF2α arm of unfolded protein response and blocking autophagy |
2019 Nov /Cell Death Dis. |
Wang D, Zhang P, Xu X, et al. |
KRT8, PERK/eIF2α arm |
|
PERK controls bone homeostasis through the regulation of osteoclast differentiation and function. |
2020 Oct/ Cell Death Dis. |
Guo J, Ren R, Sun K, et al. |
MAPK and nuclear factor κB (NF-κB) pathways, PERK |
|
ER stress arm XBP1S plays a pivotal role in proteasome inhibition-induced bone formation. |
2020 Nov/ Stem Cell Res Ther. |
Zhang D, De Veirman K, Fan R, et al. |
IRE1α-XBP1 signaling, PERK-ATF4 pathway |
|
ER stress-induced cell death in osteoarthritic cartilage. |
2021 Feb/ Cell Signal. |
Rellmann Y, Eidhof E, Dreier R |
ER stress |
|
Progranulin, a moderator of estrogen/estrogen receptor α binding, regulates bone homeostasis through PERK/p-eIF2 signaling pathway |
2022 Aug/ J Mol Med (Berl) . |
Yuyou Yang, Naibo Feng, et al. |
PERK/p-eIF2 signaling pathway |
|
Salubrinal-mediated activation of eIF2α signaling improves oxidative stress-induced BMSCs senescence and senile osteoporosis |
25 June 2022 |
LongfeiLi, GuoqinHu, et al. |
eIF2α signaling |
As a result of an imbalance between bone resorption by osteoclasts and bone synthesis by osteoblasts, osteoporosis (OP) is manifested. Estrogen has a protective effect on osteoblast apoptosis during the development of osteoporosis. 17-estradiol (17-E2, a main estrogen) inhibits osteoblast apoptosis in response to ER stress via the Ras-ERK1/2-TFIII signaling pathway [56]. Progranulin, a moderator of estrogen/estrogen receptor α binding, also be proved can regulate bone homeostasis through the PERK/p-eIF2α signaling pathway [57]. Besides, Salubrinal's (eIF2α dephosphorylation inhibitor) improvement of senile osteoporosis is also achieved by activating eIF2α signaling [58].
Autophagy is essential in the development of cartilage. Osteoblast-specific autophagy related 7 (ATG7) conditional knockout (cKO) mice showed chondrocyte apoptosis was enhanced, whereas proliferation and differentiation declined. PERK-ATF4-CHOP axis may responsible for the symptoms caused by knockout ATG7, including decreased expression of Col10a1, MMP-13, and VEGFA and increased death of chondrocytes [59]. By influencing DDIT3/CHOP and MAPK8/JNK1/SMAD1/5/8, ATG7 deficiency can also inhibit osteoblast mineralization and increase osteoblast death [60]. In addition, autophagy-related bone formation is similarly influenced by the PERK/eIF2 arm of the UPR [61].
Comprehensive analysis of the results of two kinds of differential expression profile chips, overexpression of IRE1a and ATF6 can obviously regulate the mRNA levels in MMP-14/ADAM-9/FGFR2 pathway, which is closely associated with bone development. The relevance of MMP-2, -9, -13, -14 and -16 for bone formation was established by the mice knockout and human genetic disorders [62]. Some scholars have proposed a new approach involving skull development: MMP-14/ADAM-9/FGFR2 [62]. Fibroblast growth factor receptor (FGFR) is the main factor for proper skull intramembrane ossification. Crane facial deformities are more likely be induced by the depletion of FGFR2 gene. Metalloproteases can release FGFR2 (such as ADAM-9). In MMP-14 (-/-), ADAM-9 is up-regulated, which leads to the cleavage form of FGFR2 accumulation [62]. The results of the genome-wide expression profiling chip we made found that IRE1a may be involved in this pathway to regulate skull development. After human chondrocytes overexpressed ATF6 or IRE1a, the expression of MMP-14 decreased, and the expression of ADAM-9 and FGFR2 increased
Figure 6), which is consistent with above-mentioned research results, indicating that UPR may be involve in this pathway partly to regulating bone development.
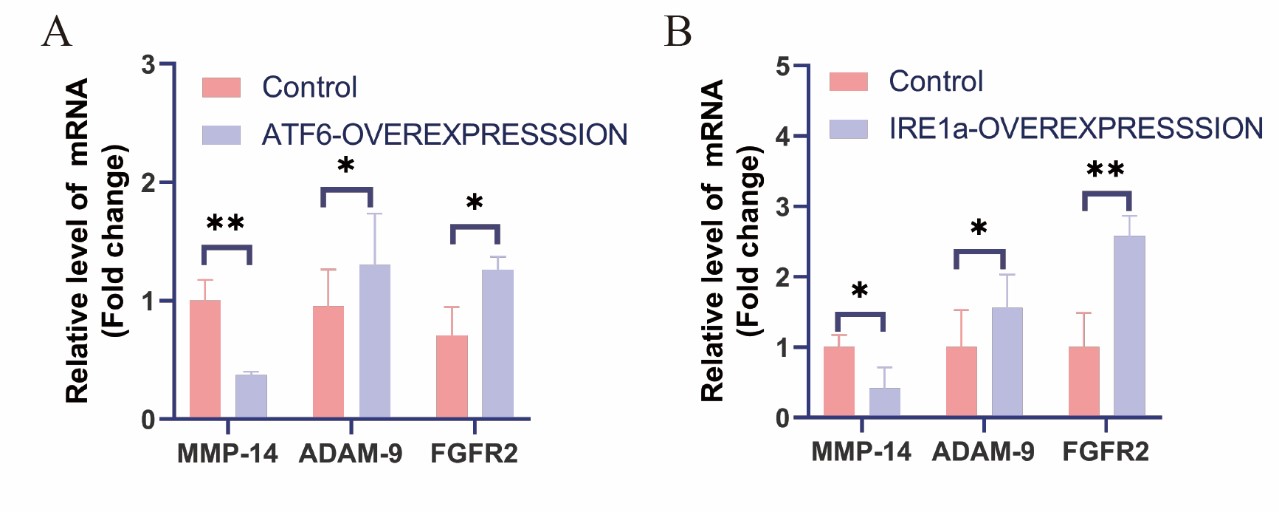
Figure 6: The differential expression profiling analysis of the common signal molecule mRNA levels of bone development and bone-related diseases genes affected by different UPR molecules. A: Quantitative analysis of MMP-14/ADAM-9/FGFR2 mRNA levels after ATF6 overexpression in human chondrocytes. B: Quantitative analysis of MMP-14/ADAM-9/FGFR2 mRNA levels after IRE1a overexpression in human chondrocytes. The error bars represe nt the SEM. *p<0.05, **p<0.01.
On basis of the result of XBP1S and ATF6 CTFM, we first selected CEBPB gene regulated by ATF6, and CTGF gene regulated by XBP1S to verify our results through the online software of prediction binding site, then analyzed the location of the gene of CEBPB and CTGF in chromosome (Figures 7A and 7B). Besides, the ATF6 binding sequence and the XBP1S binding sequence were analyzed by Jaspar database (http://jaspar.genereg.net/) on line (Figures 7C and 7D). The prediction results of the website also indicate that CEBPB or CTGF can be directly combined with ATF6 or XBP1S respectively, and the predicted sequence is GCATGGCGTGGCAT and AATGCCGCATCATT (Figures 7E and 7F).
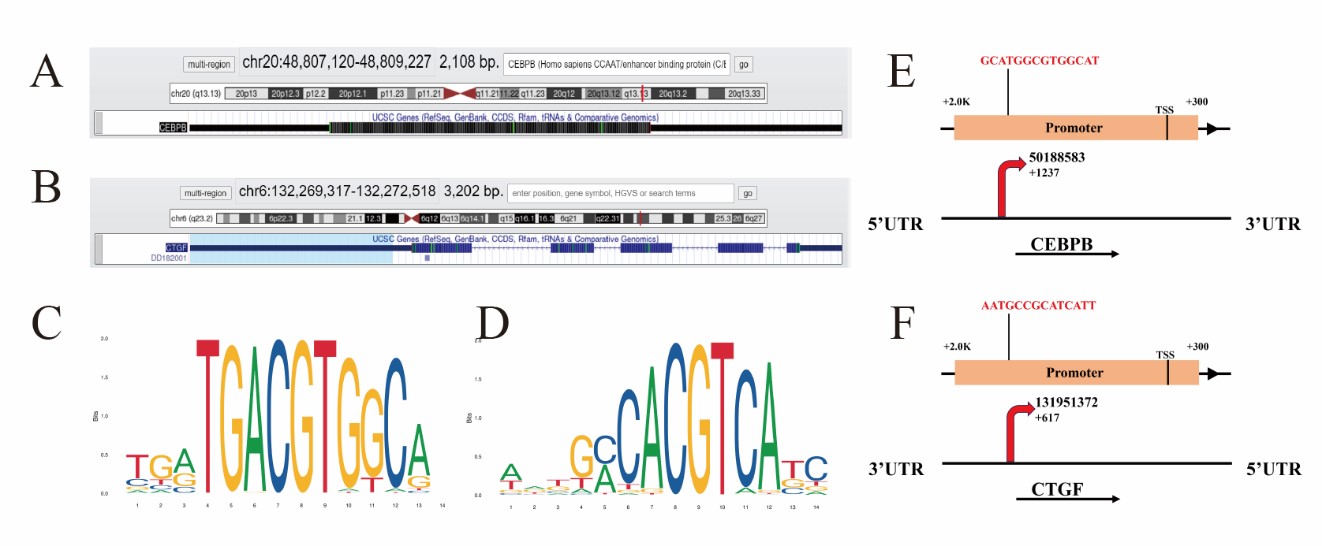
Figure 7: Predict the binding site of transcription factor ATF6 or XBP1S to the promoter region of their respective binding genes. Chromatin state annotation of the CEBPB and CTGF gene locus. The genomic region of CEBPB (transcript position: chr20, size: 2108 bp) and CTGF (transcript position: chr6: size: 3202 bp) were retrieved and are schematically represented with the indicated tracks (https://genome.ucsc.edu/) (A,B). The longest transcript was considered for each gene and the promoter region defined as 2000 bps upstream and 300 bps downstream of the Transcriptional Start Site (TSS). Each promoter sequence was analyzed using the ATF6 or XBP1S Position Weight Matrix (PWM) MA0844.1 reported in Jaspar database http://jaspar.genereg.net/ which consist of nucleotide sequence (TGATGA/GCGTGGCAT; AATGCCACG/ATCATC/T) (C, D). Their potential binding sites on the promoter region of the human CEBPB or CTGF gene with a cutoff of 80% similarity to the consensus binding sites. There are putative XBP1 binding sites on the promoter and putative XBP1 binding sites on the promoter (E, F).
Furthermore, XBP1 and ATF6 both can activate PERK-eIF2α-ATF4 signaling [38,63]. ATF4 up-regulates the expression of IRE1α mRNA in HeLa cells and mouse embryonic fibroblasts (MEF), thereby increasing the splicing rate of XBP1 [64], and a ChIP assay identified a XBP1S binding motif at the promoter region of the ATF4 gene [38]. Similarly, our microarray results also show that ATF4 can be directly combined with ATF6 and XBP1(Figure 8). All of these are proved that there exists a crosstalk between IRE1-, ATF6- and PERK-Dependent Signaling Pathways [65]. Three signal pathways of UPR obviously influenced and crosstalked each other in chondrocyte and bone metabolism.
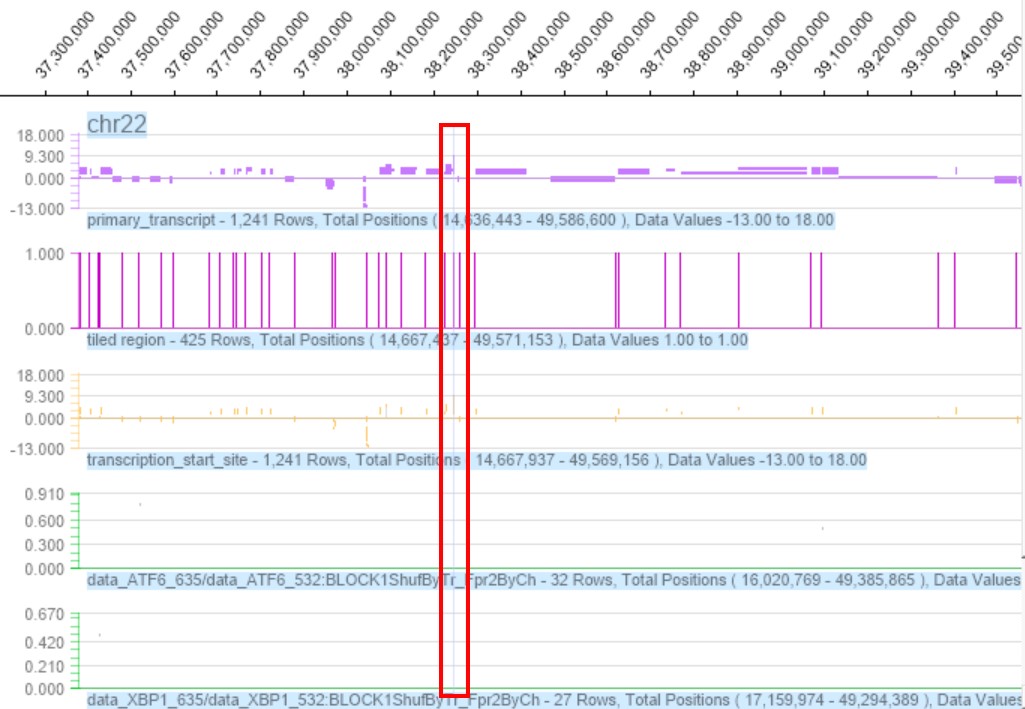
Figure 8: The ChIP-chip Microarray analysis of the promoter region of the transcription factor ATF6 and XBP1 binding ATF4 gene in chondrocytes.
PERK has garnered wide interest in its function in bone formation and remodeling. The knockout mouse has shown serious osteopenia which indicates that PERK is intimately associated with bone metabolism [66]. Mutations in the gene EIF2AK3 (encoding PERK) may be caused to induce a skeletal dysplasia and a delay in growth of Wolcott-Rallison syndrome. In addition, the EIF2AK3 haplotype gene is correlated with low bone mineral density in humans [67,68]. These all indicate the potential role of PERK in bone homeostasis. PERK inhibition or silencing have a significantly negatively effects on osteoclast differentiation, and accompanied by the down-regulation of osteoclast marker genes [66], such as MMP9 and Cathepsin K. EIF2AK3-/- Mice bone tissue expression levels are low compared to wild-type mouse with transcripted levels of osteoblast marker (including alkaline phosphatase, col I, Ocn and bone salival protein). In contrast, in the case of the EIF2AK3 and wild type mice, there is no substantial difference in osteopontin transcription (an early osteoblast differential marker) that shows that PERK is necessary for osteoblast maturation or late differentiation [69].
In the formation of skeletons, VEGF plays a vital function. Osteoblast-derived VEGF controls osteoblastogenesis and bone marrow adipogenesis, thereby stimulating osteoclast differentiation. VEGF seems to manage the differentiation of MSCs via activating the RUNX2 transcription factor and suppressing PParα2 [70]. ATF4 also promotes bone angiogenesis through its activity in osteoclasts and bone breakdown, by encouraging VEGF release from the Bone Matrix [71]. Moreover, activating ATF6 and PERK helps via favorably regulating mTORC2-mediated phospha-phosphorylation of AKT on Ser473 to the survival impact of VEGF on endothelial cells (ECs) [72,73].
Additionally, the activation of UPR was associated to cytokine dysregulation, which promotes the synthesis of lL-23, IFN-β and lL-1α and can trigger IL-23/IL-17 axis [74].The IL-17 receptor family consists of IL-17RA to IL-17RE [75], binding the heterodimer IL-17RA/IL-17RC is the most popular IL-17 signaling system. IL-17RA/IL-17RC may mediate the osteogenesis effect and IL-17RB, D and E can mediate the osteo-inhibitory effect [76-78]. Our Chip-chip transcription factor Microarray data reveal furthermore that ATF6 can directly bind to CEBPB, MAPK1, SFRS1 or MAPK13 in the IL-17 signaling pathway to directly regulate it, and also is directly engaged in regulating the VEGF signaling pathway, which may be the link between ATF6 and PERK/ATF4 pathway.
The PERK-eIf2α signal is necessary for normal bone development [79]. Salubrinal inhibits the dephosphorylation of eIf2α and increases the differentiation of osteoblasts. Regulating endoplasmic reticulum stress through eIF2α and ATF4 may be a good anti-osteoporosis system [80,81]. In diabetic patients, decreased insulin and high blood sugar lead to low bone mineral density (BMD), which impairs bone formation. In addition, diabetes can induce the expression of CHOP in osteoblasts, leading to the progression of apoptosis [82,83]. The equilibrium of osteoblasts and osteoclasts is therefore disrupted and causes diabetic osteoporosis to develop. For osteoblast maturation, the IRE1α-XBP1 route is critical to the creation of bone and the resorption of the bone under pathological circumstances [8]. ER molecular chaperones such BiP and protein disulfide isomerase (PDI) have been decreased among osteoblasts in individuals with osteoporosis(OP) [84]. These research shows the relevance for the prevention and treatment of osteoporosis of ER stress.
Furthermore, studies in the fibroblast-like synoviocytes and macrophage of rheumatoid arthritis (RA) patients demonstrated a substantial increase in activation of the IRE1/XBP1 axis [85]. IRE1α activation increases the generation of pro-inflammatory cytokines in macrophages and neutrophils mediated by the Toll-like receptor (TLR) [86]. Recently, several scientists have also hypothesized ER stress to induce inflammation by stabilization cytokine mRNA through the IRE1 RIDD activity [87]. In RA patients, RNase activity is enhanced compared with healthy persons by IRE1 in peripheral mononuclear blood cell (PBMC). Transcriptional modifications to downstream IRE1 objectives (particularly XBP1S) may therefore provide new chances to improve present diagnostic indicators and RA therapy choices [88]. The ATF6 gene is up-regulated in the macrophages of RA patients [89]. Moreover, the cleaved ATF6 form can be boosted by TNF, while proteasome or autophagy suppression can inhibit the process [90]. ATF6 may be related to the acute phase reaction and is responsible for activating a series of inflammatory mediators [89,91,92]. In synovial tissues and macrophages of RA patients the expression levels of EIF2AK3 gene and phosphorylated eIF2α are increased [89,93]. PERK/eIF2α may stimulate the NF-κB pathway to promote RA pathological inflammation [94]. In the PBMC of RA patients is also raised the level of expression of GADD34, the downstream target of the PERK pathway and is associated to the production of pro-inflammatory cytokines [85,95].
Studies have regularly been carried out in patients with RA or OA concerning the involvement of IL-17 in the articular environment. IL-17A operates on cell chondrocytes through the activation of inducible synthase of nitrogen oxide (iNOS), COX2 or cartilage degradation-related secretions IL-6. IL-17A also inhibits the synthesis of proteoglycan, increases NO generation, and works in combination with TNF alpha to kill cartilage matrix [96-98]. Additional studies have shown that IL-17F increases cartilage breakdown by boosting collagenase expression (MMP-1 and MMP-13) and stromelysin-1 (MMP-3) as well as by reducing inhibitor expression (TIMP-2 and TIMP-4) or ECM component (type II collagen, aggrecan) [99,100]. Our work also shows that overexpression IRE1a can increase collagenase expression (MMP-2, MMP-3, MMP-14). Moreover, IL-17A stimulates human chondrocytes to produce a few chemokines (CXCL1, IL-8 and CCL2), which also causes MMPs or iNOS secretions. IL-17A suppresses human MSC's Chondrogenesis, inhibiting the action of protein-kinasis A (PKA) and SOX9 phosphorylation, under inflammatory circumstances [101-103].
Conclusions
In the past decade, research has demonstrated that UPR is a critical regulator for bone growth and homeostasis. The UPR has been used in the skeletal system to normalize ER hemostasis under ER-stress, but also actively regulates cell differentiation and maturation. We do not yet completely understand how UPR promotes the pathogenesis of bone disorders, including OA, OP, and RA, as well as the physiologic function of UPR in bone formation and homeostasis. However, it is connected to some kinds of bone disorders due to overactivated UPR signaling. The three UPR pathways therefore are the main objective of research in such illnesses. Obviously, our research analyzes and predicts those UPR signaling molecules that affect cell fate through regulating ER Stress, autophagy, ROS in skeleton development and associated diseases. These molecules have certain research prospects and value for the diagnosis and treatment of bone related disease. Deeper knowledge of the link between bone homeostasis and UPR therefore provides crucial biological insights on bone metabolism and forms the basis of bone disease therapeutic intervention.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 82272550, No.81871769).
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conceptualization, Yiming Pan and Fengjin Guo; Data curation, Yiming Pan, and Fengjin Guo; Funding acquisition, Fengjin Guo; Investigation, Yiming Pan, and Fengjin Guo; Methodology, Yiming Pan, Kaiwen Liu, Yuanlan Ye, Mengtian Fan, Xiaoli Li, and Fengjin Guo; Software, Yiming Pan, Fengjin Guo; Supervision, Fengjin Guo; Writing – original draft, Yiming Pan and Fengjin Guo; Writing – review & editing, Yiming Pan and Fengjin Guo.
All authors approved the final manuscript prior to submission.
Competing Interests Statement
The authors declare that they have no conflicts of interest.
Conflicts of Interest
The authors declare no conflict of interest.
References
2. Grandjean JMD, Wiseman RL. Small molecule strategies to harness the unfolded protein response: where do we go from here? Journal of Biological Chemistry. 2020;295(46):15692-711.
3. Gonzalez-Teuber V, Albert-Gasco H, Auyeung VC, Papa FR, Mallucci GR, Hetz C. Small Molecules to Improve ER Proteostasis in Disease. Trends in Pharmacological Sciences. 2019;40(9):684-95.
4. Uddin MS, Tewari D, Sharma G, Kabir MT, Barreto GE, Bin-Jumah MN, et al. Molecular Mechanisms of ER Stress and UPR in the Pathogenesis of Alzheimer's Disease. Molecular Neurobiology. 2020;57(7):2902-19.
5. Wang M, Kaufman RJ. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nature Reviews Cancer. 2014;14(9):581-97.
6. Choi JA, Song CH. Insights Into the Role of Endoplasmic Reticulum Stress in Infectious Diseases. Frontiers in Immunology. 2019;10:3147.
7. Wei J, Sheng X, Feng D, McGrath B, Cavener DR. PERK is essential for neonatal skeletal development to regulate osteoblast proliferation and differentiation. Journal of Cellular Physiology. 2008;217(3):693-707.
8. Tohmonda T, Miyauchi Y, Ghosh R, Yoda M, Uchikawa S, Takito J, et al. The IRE1α-XBP1 pathway is essential for osteoblast differentiation through promoting transcription of Osterix. EMBO Reports. 2011;12(5):451-7.
9. Horiuchi K, Tohmonda T, Morioka H. The unfolded protein response in skeletal development and homeostasis. Cellular and Molecular Life Sciences. 2016;73(15):2851-69.
10. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science (New York, NY). 2011;334(6059):1081-6.
11. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature reviews Molecular Cell Biology. 2007;8(7):519-29.
12. Zhao N, Cao J, Xu L, Tang Q, Dobrolecki LE, Lv X, et al. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. Journal of Clinical Investigation. 2018;128(4):1283-99.
13. Xu L, Zhang W, Zhang XH-F, Chen X. Endoplasmic Reticulum Stress in Bone Metastases. 2020;10(1100).
14. Guo FJ, Jiang R, Li X, Zhang P, Han X, Liu C. Regulation of chondrocyte differentiation by IRE1α depends on its enzymatic activity. Cellular Signalling. 2014;26(9):1998-2007.
15. Lien JC, Huang CC, Lu TJ, Tseng CH, Sung PJ, Lee HZ, et al. Naphthoquinone derivative PPE8 induces endoplasmic reticulum stress in p53 null H1299 cells. Oxidative Medicine and Cellular Longevity. 2015;2015:453679.
16. Sheng W, Wang G, Tang J, Shi X, Cao R, Sun J, et al. Calreticulin promotes EMT in pancreatic cancer via mediating Ca(2+) dependent acute and chronic endoplasmic reticulum stress. Journal of Experimental & Clinical Cancer Research : CR. 2020;39(1):209.
17. Zha X, Yue Y, Dong N, Xiong S. Endoplasmic Reticulum Stress Aggravates Viral Myocarditis by Raising Inflammation Through the IRE1-Associated NF-κB Pathway. The Canadian Journal of Cardiology. 2015;31(8):1032-40.
18. So JS. Roles of Endoplasmic Reticulum Stress in Immune Responses. Molecules and Cells. 2018;41(8):705-16.
19. Hetz C, Papa FR. The Unfolded Protein Response and Cell Fate Control. Molecular Cell. 2018;69(2):169-81.
20. Cameron TL, Gresshoff IL, Bell KM, Piróg KA, Sampurno L, Hartley CL, et al. Cartilage-specific ablation of XBP1 signaling in mouse results in a chondrodysplasia characterized by reduced chondrocyte proliferation and delayed cartilage maturation and mineralization. Osteoarthritis and Cartilage. 2015;23(4):661-70.
21. Huang R, Hui Z, Wei S, Li D, Li W, Daping W, et al. IRE1 signaling regulates chondrocyte apoptosis and death fate in the osteoarthritis. Journal of Cellular Physiology. 2021.
22. Guo FJ, Xiong Z, Han X, Liu C, Liu Y, Jiang R, et al. XBP1S, a BMP2-inducible transcription factor, accelerates endochondral bone growth by activating GEP growth factor. Journal of Cellular and Molecular Medicine. 2014;18(6):1157-71.
23. Guo FJ, Jiang R, Xiong Z, Xia F, Li M, Chen L, et al. IRE1a constitutes a negative feedback loop with BMP2 and acts as a novel mediator in modulating osteogenic differentiation. Cell Death & Disease. 2014;5(5):e1239.
24. Piróg KA, Dennis EP, Hartley CL, Jackson RM, Soul J, Schwartz JM, et al. XBP1 signalling is essential for alleviating mutant protein aggregation in ER-stress related skeletal disease. PLoS Genetics. 2019;15(7):e1008215.
25. Li X, Yang Y, Liang L, Fan M, Li X, Feng N, et al. Effect Of XBP1 Deficiency In Cartilage On The Regulatory Network Of LncRNA/circRNA-miRNA-mRNA. International Journal of Biological Sciences. 2022;18(1):315-30.
26. Sharma L. Osteoarthritis of the Knee. The New England Journal of Medicine. 2021;384(1):51-9.
27. Zheng W, Li X, Liu D, Li J, Yang S, Gao Z, et al. Mechanical loading mitigates osteoarthritis symptoms by regulating endoplasmic reticulum stress and autophagy. FASEB Journal : official publication of the Federation of American Societies for Experimental Biology. 2019;33(3):4077-88.
28. Liu Z, Lv Y, Zhao N, Guan G, Wang J. Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death & Disease. 2015;6(7):e1822.
29. Rellmann Y, Eidhof E, Dreier R. Review: ER stress-induced cell death in osteoarthritic cartilage. Cellular Signalling. 2021;78:109880.
30. Wang J, Sun H, Fu Z, Liu M. Chondroprotective effects of alpha-lipoic acid in a rat model of osteoarthritis. Free Radical Research. 2016;50(7):767-80.
31. Wen T, Xue P, Ying J, Cheng S, Liu Y, Ruan D. The Role of Unfolded Protein Response in Human Intervertebral Disc Degeneration: Perk and IRE1-α as Two Potential Therapeutic Targets. Oxidative Medicine and Cellular Longevity. 2021;2021:6492879.
32. Li Z, Huang Z, Zhang H, Lu J, Wei Y, Yang Y, et al. IRE1-mTOR-PERK Axis Coordinates Autophagy and ER Stress-Apoptosis Induced by P2X7-Mediated Ca(2+) Influx in Osteoarthritis. Frontiers in Cell and Developmental Biology. 2021;9:695041.
33. Kung LHW, Mullan L, Soul J, Wang P, Mori K, Bateman JF, et al. Cartilage endoplasmic reticulum stress may influence the onset but not the progression of experimental osteoarthritis. Arthritis Research & Therapy. 2019;21(1):206.
34. Briggs MD, Dennis EP, Dietmar HF, Pirog KA. New developments in chondrocyte ER stress and related diseases. F1000Research. 2020;9.
35. Guo FJ, Xiong Z, Lu X, Ye M, Han X, Jiang R. ATF6 upregulates XBP1S and inhibits ER stress-mediated apoptosis in osteoarthritis cartilage. Cellular Signalling. 2014;26(2):332-42.
36. Cameron TL, Bell KM, Gresshoff IL, Sampurno L, Mullan L, Ermann J, et al. XBP1-Independent UPR Pathways Suppress C/EBP-β Mediated Chondrocyte Differentiation in ER-Stress Related Skeletal Disease. PLoS Genetics. 2015;11(9):e1005505.
37. Tohmonda T, Chiba K, Toyama Y, Horiuchi K. Unfolded protein response mediator, the IRE1α-XBP1 pathway is involved in osteoblast differentiation. Arthritis Research & Therapy. 2012;14(1):P70.
38. Zhang D, De Veirman K, Fan R, Jian Q, Zhang Y, Lei L, et al. ER stress arm XBP1S plays a pivotal role in proteasome inhibition-induced bone formation. Stem Cell Research & Therapy. 2020;11(1):516.
39. Tohmonda T, Yoda M, Iwawaki T, Matsumoto M, Nakamura M, Mikoshiba K, et al. IRE1α/XBP1-mediated branch of the unfolded protein response regulates osteoclastogenesis. Journal of Clinical Investigation. 2015;125(8):3269-79.
40. Dominicus CS, Patel V, Chambers JE, Malzer E, Marciniak SJ. Endoplasmic Reticulum Stress Signalling During Development. In: Clarke R, editor. The Unfolded Protein Response in Cancer. Cham: Springer International Publishing; 2019. p. 17-47.
41. Tohmonda T, Yoda M, Mizuochi H, Morioka H, Matsumoto M, Urano F, et al. The IRE1α-XBP1 pathway positively regulates parathyroid hormone (PTH)/PTH-related peptide receptor expression and is involved in pth-induced osteoclastogenesis. Journal of Biological Chemistry. 2013;288(3):1691-5.
42. Hillary RF, FitzGerald U. A lifetime of stress: ATF6 in development and homeostasis. Journal of Biomedical Science. 2018;25(1):48.
43. Guo F, Han X, Wu Z, Cheng Z, Hu Q, Zhao Y, et al. ATF6a, a RUNX2-activable transcription factor, is a new regulator of chondrocyte hypertrophy. Journal of Cell Science. 2016;129(4):717-28.
44. Nundlall S, Rajpar MH, Bell PA, Clowes C, Zeeff LA, Gardner B, et al. An unfolded protein response is the initial cellular response to the expression of mutant matrilin-3 in a mouse model of multiple epiphyseal dysplasia. Cell Stress & Chaperones. 2010;15(6):835-49.
45. Jang WG, Kim EJ, Kim DK, Ryoo HM, Lee KB, Kim SH, et al. BMP2 protein regulates osteocalcin expression via RUNX2-mediated Atf6 gene transcription. Journal of Biological Chemistry. 2012;287(2):905-15.
46. Kim JW, Choi H, Jeong BC, Oh SH, Hur SW, Lee BN, et al. Transcriptional factor ATF6 is involved in odontoblastic differentiation. Journal of Dental Research. 2014;93(5):483-9.
47. Ma M, Li H, Wang P, Yang W, Mi R, Zhuang J, et al. ATF6 aggravates angiogenesis-osteogenesis coupling during ankylosing spondylitis by mediating FGF2 expression in chondrocytes. iScience. 2021;24(7):102791.
48. Zheng C, Lin X, Xu X, Wang C, Zhou J, Gao B, et al. Suppressing UPR-dependent overactivation of FGFR3 signaling ameliorates SLC26A2-deficient chondrodysplasias. Ebiomedicine. 2019;40:695-709.
49. Forouhan M, Mori K, Boot-Handford RP. Paradoxical roles of ATF6α and ATF6β in modulating disease severity caused by mutations in collagen X. Matrix Biology. 2018;70:50-71.
50. Liu DD, Zhang BL, Yang JB, Zhou K. Celastrol ameliorates endoplasmic stress-mediated apoptosis of osteoarthritis via regulating ATF-6/CHOP signalling pathway. The Journal of Pharmacy and Pharmacology. 2020;72(6):826-35.
51. Prasadam I, Mao X, Shi W, Crawford R, Xiao Y. Combination of MEK-ERK inhibitor and hyaluronic acid has a synergistic effect on anti-hypertrophic and pro-chondrogenic activities in osteoarthritis treatment. Journal of Molecular Medicine (Berlin, Germany). 2013;91(3):369-80.
52. Gu Y, Chen J, Meng Z, Yao J, Ge W, Chen K, et al. Diazoxide prevents H(2)O(2)-induced chondrocyte apoptosis and cartilage degeneration in a rat model of osteoarthritis by reducing endoplasmic reticulum stress. Biomedicine & Pharmacotherapy = Biomedecine & Pharmacotherapie. 2017;95:1886-94.
53. Kang X, Yang W, Wang R, Xie T, Li H, Feng D, et al. Sirtuin-1 (SIRT1) stimulates growth-plate chondrogenesis by attenuating the PERK-eIF-2α-CHOP pathway in the unfolded protein response. Journal of Biological Chemistry. 2018;293(22):8614-25.
54. Li YH, Tardif G, Hum D, Kapoor M, Fahmi H, Pelletier JP, et al. The unfolded protein response genes in human osteoarthritic chondrocytes: PERK emerges as a potential therapeutic target. Arthritis Research & Therapy. 2016;18:172.
55. Botter SM, Glasson SS, Hopkins B, Clockaerts S, Weinans H, van Leeuwen JP, et al. ADAMTS5-/- mice have less subchondral bone changes after induction of osteoarthritis through surgical instability: implications for a link between cartilage and subchondral bone changes. Osteoarthritis and Cartilage. 2009;17(5):636-45.
56. Guo YS, Sun Z, Ma J, Cui W, Gao B, Zhang HY, et al. 17β-Estradiol inhibits ER stress-induced apoptosis through promotion of TFII-I-dependent Grp78 induction in osteoblasts. Laboratory Investigation; A Journal of Technical Methods and Pathology. 2014;94(8):906-16.
57. Yang Y, Feng N, Liang L, Jiang R, Pan Y, Geng N, et al. Progranulin, a moderator of estrogen/estrogen receptor α binding, regulates bone homeostasis through PERK/p-eIF2 signaling pathway. Journal of Molecular Medicine (Berlin, Germany). 2022;100(8):1191-207.
58. Li L, Hu G, Xie R, Yang J, Shi X, Jia Z, et al. Salubrinal-mediated activation of eIF2α signaling improves oxidative stress-induced BMSCs senescence and senile osteoporosis. Biochemical and Biophysical Research Communications. 2022;610:70-6.
59. Kang X, Yang W, Feng D, Jin X, Ma Z, Qian Z, et al. Cartilage-Specific Autophagy Deficiency Promotes ER Stress and Impairs Chondrogenesis in PERK-ATF4-CHOP-Dependent Manner. Journal of Bone and Mineral Research : The Official Journal of the American Society for Bone and Mineral Research. 2017;32(10):2128-41.
60. Li H, Li D, Ma Z, Qian Z, Kang X, Jin X, et al. Defective autophagy in osteoblasts induces endoplasmic reticulum stress and causes remarkable bone loss. Autophagy. 2018;14(10):1726-41.
61. Wang D, Zhang P, Xu X, Wang J, Wang D, Peng P, et al. Knockdown of cytokeratin 8 overcomes chemoresistance of chordoma cells by aggravating endoplasmic reticulum stress through PERK/eIF2α arm of unfolded protein response and blocking autophagy. Cell Death & Disease. 2019;10(12):887.
62. Paiva KBS, Granjeiro JM. Bone tissue remodeling and development: Focus on matrix metalloproteinase functions. Archives of Biochemistry and Biophysics. 2014;561:74-87.
63. Spaan CN, Smit WL, van Lidth de Jeude JF, Meijer BJ, Muncan V, van den Brink GR, et al. Expression of UPR effector proteins ATF6 and XBP1 reduce colorectal cancer cell proliferation and stemness by activating PERK signaling. Cell Death & Disease. 2019;10(7):490.
64. Tsuru A, Imai Y, Saito M, Kohno K. Novel mechanism of enhancing IRE1α-XBP1 signalling via the PERK-ATF4 pathway. Scientific Reports. 2016;6:24217.
65. Siwecka N, Rozpędek-Kamińska W, Wawrzynkiewicz A, Pytel D, Diehl JA, Majsterek I. The Structure, Activation and Signaling of IRE1 and Its Role in Determining Cell Fate. Biomedicines. 2021;9(2).
66. Guo J, Ren R, Sun K, Yao X, Lin J, Wang G, et al. PERK controls bone homeostasis through the regulation of osteoclast differentiation and function. Cell Death & Disease. 2020;11(10):847.
67. Horiuchi K, Tohmonda T, Morioka H. The unfolded protein response in skeletal development and homeostasis. Cellular and Molecular Life Sciences : CMLS. 2016;73(15):2851-69.
68. Liu J, Hoppman N, O'Connell JR, Wang H, Streeten EA, McLenithan JC, et al. A functional haplotype in EIF2AK3, an ER stress sensor, is associated with lower bone mineral density. Journal of Bone and Mineral Research : The Official Journal of the American Society for Bone and Mineral Research. 2012;27(2):331-41.
69. Saito A, Ochiai K, Kondo S, Tsumagari K, Murakami T, Cavener DR, et al. Endoplasmic reticulum stress response mediated by the PERK-eIF2(alpha)-ATF4 pathway is involved in osteoblast differentiation induced by BMP2. Journal of Biological Chemistry. 2011;286(6):4809-18.
70. Liu Y, Olsen BR. Distinct VEGF functions during bone development and homeostasis. Archivum immunologiae et Therapiae Experimentalis. 2014;62(5):363-8.
71. Zhu K, Jiao H, Li S, Cao H, Galson DL, Zhao Z, et al. ATF4 promotes bone angiogenesis by increasing VEGF expression and release in the bone environment. Journal of Bone and Mineral Research : The Official Journal of the American Society for Bone and Mineral Research. 2013;28(9):1870-84.
72. Karali E, Bellou S, Stellas D, Klinakis A, Murphy C, Fotsis T. VEGF Signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress. Molecular Cell. 2014;54(4):559-72.
73. Rellmann Y, Gronau I, Hansen U, Dreier R. 4-Phenylbutyric Acid Reduces Endoplasmic Reticulum Stress in Chondrocytes That Is Caused by Loss of the Protein Disulfide Isomerase ERp57. Oxidative Medicine and Cellular Longevity. 2019;2019:6404035.
74. Colbert RA, Tran TM, Layh-Schmitt G. HLA-B27 misfolding and ankylosing spondylitis. Molecular Immunology. 2014;57(1):44-51.
75. Gaffen SL. Structure and signalling in the IL-17 receptor family. Nature Reviews Immunology. 2009;9(8):556-67.
76. Osta B, Lavocat F, Eljaafari A, Miossec P. Effects of Interleukin-17A on Osteogenic Differentiation of Isolated Human Mesenchymal Stem Cells. Frontiers in Immunology. 2014;5:425.
77. Shaw AT, Maeda Y, Gravallese EM. IL-17A deficiency promotes periosteal bone formation in a model of inflammatory arthritis. Arthritis Research & Therapy. 2016;18(1):104.
78. Kim HJ, Seo SJ, Kim JY, Kim YG, Lee Y. IL-17 promotes osteoblast differentiation, bone regeneration, and remodeling in mice. Biochemical and Biophysical Research Communications. 2020;524(4):1044-50.
79. Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Molecular Cell. 2001;7(6):1153-63.
80. He L, Lee J, Jang JH, Sakchaisri K, Hwang J, Cha-Molstad HJ, et al. Osteoporosis regulation by salubrinal through eIF2α mediated differentiation of osteoclast and osteoblast. Cellular Signalling. 2013;25(2):552-60.
81. Li J, Yang S, Li X, Liu D, Wang Z, Guo J, et al. Role of endoplasmic reticulum stress in disuse osteoporosis. Bone. 2017;97:2-14.
82. Norris R, Parker M. Diabetes mellitus and hip fracture: a study of 5966 cases. Injury. 2011;42(11):1313-6.
83. Liu W, Zhu X, Wang Q, Wang L. Hyperglycemia induces endoplasmic reticulum stress-dependent CHOP expression in osteoblasts. Experimental and Therapeutic Medicine. 2013;5(5):1289-92.
84. Mahdi AA, Rizvi SH, Parveen A. Role of Endoplasmic Reticulum Stress and Unfolded Protein Responses in Health and Diseases. Indian Journal of Clinical Biochemistry : IJCB. 2016;31(2):127-37.
85. Rahmati M, Moosavi MA, McDermott MF. ER Stress: A Therapeutic Target in Rheumatoid Arthritis? Trends in Pharmacological Sciences. 2018;39(7):610-23.
86. Qiu Q, Zheng Z, Chang L, Zhao YS, Tan C, Dandekar A, et al. Toll-like receptor-mediated IRE1α activation as a therapeutic target for inflammatory arthritis. The EMBO Journal. 2013;32(18):2477-90.
87. Kabala PA, Angiolilli C, Yeremenko N, Grabiec AM, Giovannone B, Pots D, et al. Endoplasmic reticulum stress cooperates with Toll-like receptor ligation in driving activation of rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Research & Therapy. 2017;19(1):207.
88. Ahmadiany M, Alavi-Samani M, Hashemi Z, Moosavi MA, Rahmati M. The Increased RNase Activity of IRE1α in PBMCs from Patients with Rheumatoid Arthritis. Advanced Pharmaceutical Bulletin. 2019;9(3):505-9.
89. Yoo SA, You S, Yoon HJ, Kim DH, Kim HS, Lee K, et al. A novel pathogenic role of the ER chaperone GRP78/BiP in rheumatoid arthritis. The Journal of Experimental Medicine. 2012;209(4):871-86.
90. Connor AM, Mahomed N, Gandhi R, Keystone EC, Berger SA. TNFα modulates protein degradation pathways in rheumatoid arthritis synovial fibroblasts. Arthritis Research & Therapy. 2012;14(2):R62.
91. Ta HM, Le TM, Ishii H, Takarada-Iemata M, Hattori T, Hashida K, et al. Atf6α deficiency suppresses microglial activation and ameliorates pathology of experimental autoimmune encephalomyelitis. Journal of Neurochemistry. 2016;139(6):1124-37.
92. Zhou F, Mei J, Han X, Li H, Yang S, Wang M, et al. Kinsenoside attenuates osteoarthritis by repolarizing macrophages through inactivating NF-κB/MAPK signaling and protecting chondrocytes. Acta Pharmaceutica Sinica B. 2019;9(5):973-85.
93. Guo J, Ren R, Sun K, He J, Shao J. PERK signaling pathway in bone metabolism: Friend or foe? Cell Proliferation. 2021;54(4):e13011.
94. Sinjari B, Pizzicannella J, D'Aurora M, Zappacosta R, Gatta V, Fontana A, et al. Curcumin/Liposome Nanotechnology as Delivery Platform for Anti-inflammatory Activities via NFkB/ERK/pERK Pathway in Human Dental Pulp Treated With 2-HydroxyEthyl MethAcrylate (HEMA). Frontiers in Physiology. 2019;10:633.
95. Park YJ, Yoo SA, Kim WU. Role of endoplasmic reticulum stress in rheumatoid arthritis pathogenesis. Journal of Korean Medical Science. 2014;29(1):2-11.
96. Faust HJ, Zhang H, Han J, Wolf MT, Jeon OH, Sadtler K, et al. IL-17 and immunologically induced senescence regulate response to injury in osteoarthritis. Journal of Clinical Investigation. 2020;130(10):5493-507.
97. Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nature Reviews Rheumatology. 2011;7(1):33-42.
98. Le Goff B, Bouvard B, Lequerre T, Lespessailles E, Marotte H, Pers YM, et al. Implication of IL-17 in Bone Loss and Structural Damage in Inflammatory Rheumatic Diseases. Mediators of Inflammation. 2019;2019:8659302.
99. Li Z, Yuan B, Pei Z, Zhang K, Ding Z, Zhu S, et al. Circ_0136474 and MMP-13 suppressed cell proliferation by competitive binding to miR-127-5p in osteoarthritis. Journal of Cellular and Molecular Medicine. 2019;23(10):6554-64.
100. Wu Z, He D, Zhao S, Wang H. IL-17A/IL-17RA promotes invasion and activates MMP-2 and MMP-9 expression via p38 MAPK signaling pathway in non-small cell lung cancer. Molecular and Cellular Biochemistry. 2019;455(1-2):195-206.
101. Mojsilović S, Jauković A, Santibañez JF, Bugarski D. Interleukin-17 and its implication in the regulation of differentiation and function of hematopoietic and mesenchymal stem cells. Mediators of Inflammation. 2015;2015:470458.
102. Malta FS, Garcia RP, Azarias JS, Ribeiro G, Miranda TS, Shibli JA, et al. Impact of hyperglycemia and treatment with metformin on ligature-induced bone loss, bone repair and expression of bone metabolism transcription factors. PLoS One. 2020;15(8):e0237660.
103. Park SW, Kim M, Brown KM, D'Agati VD, Lee HT. Paneth cell-derived interleukin-17A causes multiorgan dysfunction after hepatic ischemia and reperfusion injury. Hepatology (Baltimore, Md). 2011;53(5):1662-75.