1. Introduction
Mutational signatures, defined as characteristic patterns of somatic mutations in cancer genomes, shed light on the mutagenic mechanisms and origins of cancer. Mutational signature analysis [1–4] leverages computational techniques to identify recurring patterns within the spectrum of genetic alterations observed in tumors. Using mutational signature analysis, investigators can identify biological processes responsible for tumorigenesis. For example, Nik-Zainal et al. [5] used mutational signature analysis to identify signatures associated with the activity of APOBEC cytidine deaminases, a family of enzymes that can induce cytosine-to-uracil deamination, leading to characteristic C>T and C>G mutations at TpC dinucleotides. This signature pointed to endogenous APOBEC activity as a significant mutagenic process in the development of breast cancer. For another example, Alexandrov et al. [6] investigated mutational signatures in lung cancers occurring in smokers and found a strong prevalence of a signature characterized by C>A transversions. This signature was experimentally linked to benzo[a]pyrene, a carcinogen present in tobacco smoke, providing evidence that tobacco-induced DNA damage is a driving mutational process in lung tumorigenesis. In addition to their role in the elucidation of cancer etiology, mutational signatures can provide therapeutic and prognostic insights [4,7], as documented in a recent review by Brady 2022 [8]. For example, mutational signatures of homologous recombination deficiency are predictive of PARP inhibitor efficacy, mutational signatures indicating mismatch repair deficiency are directly correlated with response to immune checkpoint blockade, while APOBEC-related signatures are predictive of ATR inhibitor efficacy.
The development of computational methods for the estimation of mutational signatures in increasingly complex data sets is an important area in cancer research. A 2022 review identified 14 methods for the estimation of mutational signatures de novo [9]. Since somatic mutation data are often collected as part of different studies, ranging from large-scale sequencing efforts [4] to correlative studies within clinical trials, [10,11] there is a growing need for multi-study methodologies that facilitate integrated analyses of multiple datasets. Important questions include, for example, identifying and removing artifacts that make it insufficient to simply merge data from different sources; discerning whether signatures identified in a study are or not the same as those previously proposed; contrasting studies that consider different subtypes or conditions, and so forth. Here, we review two recent contributions in this area, in which we endeavored to offer a rigorous and comprehensive statistical framework for mutational signature analysis over multiple datasets. Although many approaches have been introduced to summarize and interpret the results of multiple signature analyses from different studies and distinct datasets, integrated statistical models allow joint analyses of multiple datasets. Both these models offer two types of advantages: 1) an increased learning efficiency compared to analyses based on a single dataset and meta-analyses, and 2) a support for rigorous assessment of the uncertainty on the results. Finally, we highlight the type of cancer biology insight that researchers can glean from this kind of integrated analysis.
2. Methodology
2.1 Nonnegative matrix factorization for mutational signatures
To better understand how mutations arise in cancer, researchers have developed systems to classify single-base substitutions (SBSs) based on both the type of mutation and the surrounding DNA sequence. A widely used motif-based scheme [2,5,12,13] formalized a system based on the six possible substitutions (C>A, C>G, C>T, T>A, T>C, T>G), reported on the DNA strand where the original base is a pyrimidine (C or T), and the 4 nucleotides flanking the mutated base on each side. This results in I = 4´6´4 = 96 different types or “motifs” of mutations. Based on the individual counts of somatic mutations of each type, patterns can be learned using Non-negative Matrix Factorization (NMF) [14]. Considering first a single study, for each tumor j = 1, . . . , J, let mij denote the number of observed SNVs of motif i in patient j. These counts form the mutation count matrix M of dimension I ×J. Like other popular dimension reduction approaches, such as principal component analysis, NMF seeks a concise representation of this data as: M ≈ P E, where P is an I × K matrix whose column pk represents the k-th mutational signature, with pik denoting the frequency of the motif i in the signature k. In NMF, unlike other dimension reduction approaches, both P E are positive and the columns of P are not required to be orthogonal. This permit learning the activity of separate biological processes even when they have some preferred motifs in common. The K × J matrix E represents signature exposures. These quantify the contribution of each signature to the mutation counts in each tumor. This factorization expresses the mutation profile of each tumor as the sum of the contributions arising from a small number of mutational signatures, each associated with a specific pattern of nucleotide changes. This framework can also be extended to include dinucleotide mutations, indels, copy number changes, structural variants, and other mutation types by defining appropriate motifs.
In a de novo analysis, both the signatures and the exposures can be learned from the mutation count matrix. Optimization approaches proceed by minimizing ||M − P E|| with respect to P and E for a fixed rank K and norm || · ||, possibly with additional regularization. ||M − P E|| quantifies how well P and E can reconstruct M. In statistical approaches, M is seen as a random matrix governed by parameters P and E. A statistical model can reflect directly how the data are collected, for example via conditional independence assumption when patients are a random sample from a population. Statistical models can also explicitly account for measurement error, and other sources of variability [15–17]. Table 1 provides a summary, likely to be incomplete very soon, of methods to infer signatures and to learn the distribution of exposures in a population from somatic mutation counts. A comparison of signature analysis methods and software is available on the SigFitTest [18] website.
|
Topic |
Software Packages / References |
|
Early approaches using the Poisson model |
Emu [19], signer [16] |
|
Dirichlet mixture specifications |
Pmsignature [20], sigfit [21], mSigHdp [22], Compressive NMF [23], Hansen 2025 [24] |
|
Regularization methods for sparsity, stability, and automatic rank selection |
SignatureAnalyzer [25], sigLASSO [26], sparseSignatures [27], SigProfilerExtractor [9], MuSiCal [28], Compressive NMF [23] |
|
Alternative prior specifications |
Sigfit [21], BayesPowerNMF [29], BayesNMF [30] |
|
Ensembling |
MetaMutationalSigs [31] |
|
Higher resolution alphabets |
TensorSignatures [32] |
|
Efficient Bayesian Decomposition with Principled Automatic Identification of Rank |
bayesNMF [30] |
|
Joint discovery and recovery of known signatures |
SignatureAnalyzer [25], sigLASSO [26], SigProfilerExtractor [9], Grabski 2025 [17] |
|
Multi-study analyses |
Grabski 2025 [17], Hansen 2025 [24] |
|
Covariates and spatial variation |
Robinson 2019 [33], Grabski 2025 [17], Hansen 2025 [24], SigProfilerTopography [34] |
|
Dependence in Mutation-Type Probabilities |
Lang 2025 [35] |
|
Autoencoder-based signatures |
MUSE-XAE [36] |
|
Other available software. Most of these toolboxes provide “one-stop” solutions with extensive functionality. |
COSMIC [37] (includingSigProfiler), SomaticSignatures [38], MutSpec [39], mSignatureDB [40], MuSiCa [41], BayesNMF [30], Signal [42], Sigflow [43], MutSignatures [44], mmsig [45], muscatk [46], MutationalPatterns [47], SigProfilerAssignment [48] |
2.2 Multi-study analysis
In the context of mutational signature analysis, data integration plays a crucial role in uncovering biologically meaningful patterns that may be obscured in single-source or single-cohort analyses. There are two complementary types of integration problems: 1) Multi-study integration, where somatic mutation data, summarized as mutation motif counts, are collected across multiple independent studies, cohorts, or cancer types, each measuring the same set of mutational features; and 2) Multi-modal (or multi-platform) integration, where different types of complementary data, such as mutation signatures, gene expression, copy number alterations, or clinical covariates, are measured on a set of tumor samples or patients.
The focus of this article is multi-study analysis, whose primary goal is to decompose mutational counts into shared signatures that recur across studies and study-specific components that reflect unique processes, such as population-specific exposures. For example, integrating SNV data from multiple cancer cohorts can enhance the recovery of robust mutational processes, such as aging, while also isolating noise or signals unique to a given study.
It is important to distinguish multi-study analysis from Multi-modal integration which, in contrast, seeks to jointly model heterogeneous data modalities that provide complementary views of tumor biology on the same tumor. For example, for a tumor sample, investigators may have measurements of mutational signature exposures, transcriptomic profiles, methylation states, and radiological [49]. Mathematically, in a multi-study analysis the multiple matrices share common labels of the variables or rows, while in multi-model analyses the multiple matrices share common labels of the columns, or subjects. Although many existing signature studies focus only on SNV, integrating additional molecular layers or clinical characteristics can reveal how mutational processes relate to phenotypic variation, treatment response, or prognosis [49,50]. Without attempting a systematic review, recent statistical methods, such as multi-omics factor analysis [51] and DIABLO [52], jointly infer latent structures across complementary data types, revealing relations between mutational processes and other tumor-specific features.
In this review, we focus on NMF methods specifically designed for multi-study analysis. Within a Bayesian modeling framework, recent advances have addressed several key challenges: jointly analyzing multiple studies or conditions; integrating patient-level covariates to account for individual heterogeneity; incorporating previously known signatures into probabilistic models; and learning concise representations of mutational signature exposures at the individual level. Two models have been developed to address non-negative matrix factorization in the multi-study framework: Multi- Study Non-Negative Matrix Factorization [17] and the Bayesian Probit Multi-Study Non-negative Matrix Factorization [24].
2.3 Bayesian Models
2.3.1 Bayesian NMF: We now extend the setting of Section 2.1 to the case of S studies, each comprising Js observations (e.g. tumors) and providing the count matrix Ms of dimension I × Js, for s = 1, · · ·, S. In Multi-Study Non-Negative Matrix Factorization [17], a key feature is that the set of active mutational processes is specific to the study, but if process k is shared by multiple studies, the corresponding signature pk remains the same. To achieve this, the mutational counts of study s are decomposed as:

for s = 1, . . . S. Here, dimensions are in gray. The matrix P includes all signatures active in at least one study. The matrix As is a diagonal study-specific matrix of indicators that identify signatures active in study s, that is, the k-th diagonal element of As is 1 if signature k is active in study s, and 0 otherwise. Lastly, Es is the exposure matrix for the study s.
Bayesian modeling proceeds by assigning prior distributions to these matrices. Priors can be specified to be relatively vague or can incorporate information from prior studies or catalogs, as we will see later. Grabski [17] assigns conjugate Gamma priors to the elements of both the signature and exposure matrices:
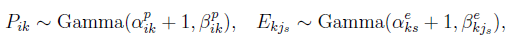
with hyperpriors placed on the shape and rate parameters to allow adaptive regularization:

The binary inclusion matrix A, which encodes study-specific signature activity, is given a Bernoulli prior Ask ∼ Bernoulli(q), allowing for sparsity in signature sharing across studies.
2.3.2 Recovery and discovery using Bayesian NMF: A key innovative use of this model is the introduction of semi-supervised recovery-discovery matrix factorization, which allows for simultaneous identification of known signatures and discovery of novel ones. The resulting method incorporates signatures from, for example, the Cosmic [37] database, while allowing for uncertainty in the precise values of the entries of Pk . To achieve this, NMF decomposition is split into two additive components: one for recovery of known signatures and another for discovery of novel study-specific signatures, such as those arising from previously unexplored tissues, exposures, treatments, or sequencing protocols. Formally, we arrange the Ms ∼ Poisson (PAsEs) structure as follows:
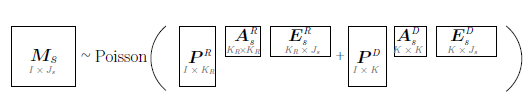
where PR represents the recovery component and contains a fixed number KR of previously proposed (“old”) signatures. Although elements of PR are previously proposed signatures, they have been estimated with varying degrees of statistical errors and occasionally might be affected by artifacts. Based on these considerations, they are assigned strong priors to leverage existing knowledge and encourage estimates close to the original proposals but also allow small departures. In contrast, PD represents the discovery component, consisting of an unknown number K of “new” signatures. The study-specific presence or absence of these signatures is encoded by binary indicator matrices AsR and AsD, for the recovery and discovery components, respectively.
2.3.3 Bi-clustering and covariate effects via sparsity in exposures: BaP Multi-NMF: Not all signatures are active in every tumor, a characteristic that is captured by positing that the exposure matrix is sparse, that is that it includes many zeros. We developed a multi-study probit mixture model (BaP Multi-NMF) to encode this sparsity [24], allowing each tumor to be represented by only a subset of signatures and allowing this subset to depend on covariates. BaP Multi-NMF accomplishes this goal by using a mixture prior on exposures, clustering exposures into one of two groups: one group denoting substantial exposure or detection of a mutational signature, and another group denoting negligible exposure or no detection. In effect, BaP Multi-NMF enforces a shrinkage effect on non-important mutational signatures. This approach enables: (a) more accurate identification of mutational signature contributions at the individual level; (b) improved estimation of signature activity prevalence within cancer types; and (c) de novo identification of interpretable patient subtypes across cancers based on their mutational profiles.
In the BaP Multi-NMF model, the mutational counts are decomposed as:

where P, as above, is the I × K signatures matrix and Es is the K × Js exposures matrix. In Hansen 2025 paper [24] the matrix Ws = diag (ws1, . . . , wsJs) is fixed and its entries are set at the total mutation counts by subject and motif, that is  . These quantities are actually unknown prior to conducting the study and in some cases (e.g., extreme genomic instability) can be informative of signature activity. Thus, this is a limitation of this specification, introduced for computational efficiency.
. These quantities are actually unknown prior to conducting the study and in some cases (e.g., extreme genomic instability) can be informative of signature activity. Thus, this is a limitation of this specification, introduced for computational efficiency.
In contrast to the Grabski 2025 paper [17], this model assumes that the columns of the signature and exposure matrices are normalized to sum to one and assigns them Dirichlet priors:
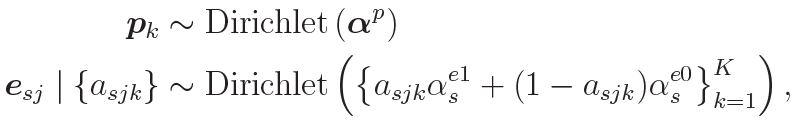
where Pk is the kth column of the signature matrix P and the vector  is set to ( , . . . , ) for some
is set to ( , . . . , ) for some  . In addition, esj is the jth column of the exposure matrix Es for study s and
. In addition, esj is the jth column of the exposure matrix Es for study s and  and
and  such that
such that  is much greater than
is much greater than  so that two components well approximate inclusion and exclusion of exposure esjk.
so that two components well approximate inclusion and exclusion of exposure esjk.
This hierarchical model incorporates covariate information xsj into the signature process via a probit hyper-prior on Es, allowing the model to flexibly modulate exposure distributions based on sample-level features. To this end, the variable  is important for the interpretation of the model’s results. It is a binary variable and indicates whether the signature k is active specifically in sample j of study s, depending on both mutation counts and patient characteristics. This is achieved by modeling the latent variable asjk*
is important for the interpretation of the model’s results. It is a binary variable and indicates whether the signature k is active specifically in sample j of study s, depending on both mutation counts and patient characteristics. This is achieved by modeling the latent variable asjk* using probit regression:
using probit regression:
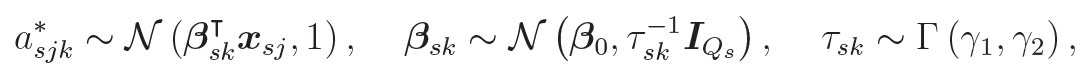
where 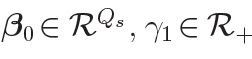 , and
, and  are fixed hyperparameters.
are fixed hyperparameters.
Estimation of signatures and analysis of the influence of covariates, such as cancer treatments or age, on the number of mutations associated to each signature have been approached as two separate tasks. The Bayesian NMF models that we described allow users to estimate signatures and investigate the role of covariates by incorporating covariate effects. In particular, the Probit model estimates which signatures contributed to mutation counts. Various forms of relationships between covariates and mutations can be explored through the rich catalog of links from the generalized linear models literature. For example, to obtain a re-scaling effect of covariates on the individual exposures, one can (dropping the study subscript for a moment) model the expectation of exposures ekj to be a function of the covariates, like  . We used these link functions to investigate relationships between tumor-level exposures and covariates and to assess whether factors such as sex, smoking status, or inherited susceptibility influence the individual mutation profile [17,24]. Although previous studies have correlated estimated exposures with patient characteristics [33,53,54], the study of joint Bayesian models has shown that covariates can also be leveraged to improve the estimation of mutational signatures.
. We used these link functions to investigate relationships between tumor-level exposures and covariates and to assess whether factors such as sex, smoking status, or inherited susceptibility influence the individual mutation profile [17,24]. Although previous studies have correlated estimated exposures with patient characteristics [33,53,54], the study of joint Bayesian models has shown that covariates can also be leveraged to improve the estimation of mutational signatures.
3. Early Onset Breast Cancer Signatures and Smoking
We applied our multi-study NMF approach to study the role of mutational signatures in early-onset breast cancer [17]. Such cases are associated with worsened survival and more aggressive presentation, yet much remains unknown about their underlying mutational processes. Previous work has used conventional, single-study methods to compare mutational signatures across age [55], but a multi-study approach allows us to more systematically discover the shared and unique mutational processes underlying these age groups. Moreover, our approach naturally enables the inclusion of data from multiple cohorts, which improves the power to detect subtle effects.
Specifically, we conducted an analysis considering breast tumors spanning multiple age groups from two different sources, TCGA and PCAWG. In our analysis a “study” is a combination of age group (20–29, 30–39 or 40–49) and source (TCGA and PCAWG), allowing us to address both 6 biological differences between age groups, and replicability of signature discovery across sources.
Our multi-study methods are particularly well-suited for scenarios like these, where there are substantial technology-driven differences between the two sources and sometimes small sample sizes, e.g. with just seven tumors in the TCGA 20–29 group. Using our recovery-discovery approach, we ultimately found a total of 41 signatures including many that have been previously reported to play an important role in breast cancer, such as the canonical signature SBS3 that represents defective homologous recombination DNA damage repair.
Interestingly, however, the youngest group (TCGA 20-29) lacked SBS3, along with some other traditional breast cancer mutational signatures. Instead, there were several signatures unique to this group that pertain to environmental exposures, including the canonical smoking signature SBS4. This suggests that, uniquely in this age group, environmental and lifestyle factors may play a greater role than some of the common mutational processes, e.g. those associated with germline BRCA1 and BRCA2 mutations, that typically lead to breast cancer development. While caution in interpretation is required given the small sample size, this analysis highlights our ability to extract signal in highly challenging settings where single-study approaches would be much more limited.
4. Identifying Patient Clusters Across Cancer Types
Accurate identification of clusters of patients that share mutational mechanisms can inform precision medicine. To illustrate BaP Multi-NMF, we analyzed tumor DNA from seven cancer types [24] and identified clusters of both signatures and patients (see Figure 1). At the level of an individual tumor, the parameter asjr defined a binary” mutational profile” listing the mutational signatures that are active in that tumor. This results in a useful latent space that contains information about the relationships between mutational profiles and signatures.
We revisit this analysis in Figure 1. In this heatmap, each entry represents the posterior mean of asjr, with rows representing profiles and columns representing signatures. The profiles and signatures are independently clustered using Ward’s minimum variance method based on the Manhattan distance. The results are consistent with our earlier findings in Hansen 2025 [24].
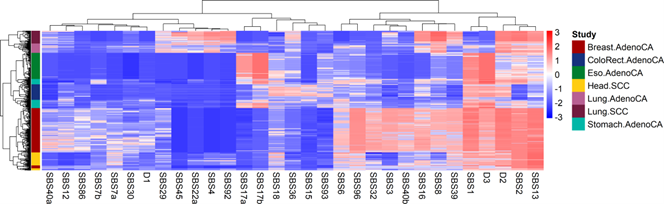
Figure 1. Multi-cancer bi-clustering analysis: Heatmap visualizing inclusion probabilities of mutational signatures across all samples in the 7 cancer types considered. In this matrix, each row corresponds to a mutational profile, each column corresponds to a mutational signature, and the color of the heatmap is determined by the posterior mean of asjr* . Mutational profiles and mutational signatures are both clustered using Ward’s minimum variance method with the Manhattan distance metric.
. Mutational profiles and mutational signatures are both clustered using Ward’s minimum variance method with the Manhattan distance metric.
Our recovery-discovery analysis identifies known mutational signatures as well as three novel ones labeled D1, D2, and D3. The dendrogram at the top suggests that signatures form two large clusters with some indication of further partitioning into subclusters. A cohesive subcluster includes mutational signatures associated with tobacco use, SBS4, SBS2, SBS92 together with SBS22a and SBS45. These are active in most lung cancers, including adenocarcinomas and squamous cell carcinomas (SCC). Also, SBS93, a signature of unknown etiology, correlates strongly with SBS3 (Defective homologous recombination-based DNA damage repair) and other well understood signatures in the cluster ranging from SBS6 to SBS39 of our heatmap.
BaP Multi-NMF can also answer questions about how mutational profiles of different cancer types relate to each other. We find three main clusters: one that contains breast and head cancers, one containing stomach, colorectal, and esophagus cancers, and one containing lung adenocarcinoma and lung squamous cell carcinoma. Each cluster can be divided into subgroups defined by cancer type, with some notable exceptions. First, we found a small group of breast cancers that were more similar to head cancers than the majority of breast cancers. This result appears to be driven by lower exposure to mutational signatures SBS40a, SBS12, and SBS3 and, more generally, signatures in the cluster ranging from SBS6 to SBS39. Another interesting finding is a subgroup of stomach cancers that clustered with esophageal cancers, as a result of higher exposures to SBS17a compared to other stomach cancers.
Overall, these results suggest that the approach taken in my BaP Multi-NMF not only has the potential to estimate sparse exposures using subject-level indicators but can also generate an enriched space which contains important information regarding the latent structure of mutational signatures and profiles.
5. Software
R code implementing the methods described in “Bayesian Multi-Study Non-Negative Matrix Factorization for Mutational Signatures” [17] and “Bayesian Probit Multi-Study Non-negative Matrix Factorization for Mutational Signatures” [24] is available in the respective GitHub repositories referenced in those publications.
6. Funding
B.H. was supported by the US National Institutes of Health, under grants NIGMS/NIH COBRE CBHD P20GM109035and. R.D.V. was supported by the US National Institutes of Health, under grants NIGMS/NIH COBRE CBHD P20GM109035, and 5R01CA262710. L.T and G.P. were supported by the U.S.A. National Institutes of Health, through grants 5R01CA262710 and 5R01CA240299 (L.T. only) and by the U.S.A. National Science Foundation through grant DMS 2113707. I.N.G. is the Kenneth G. Langone Quantitative Biology Fellow of the Damon Runyon Cancer Research Foundation (DRQ-21-24).
7. Conflicts of Interest
While he does not consider any of these activities to be in conflict with the research in this article, Giovanni Parmigiani wishes to disclose that he holds equity in Phaeno Biotechnologies and currently consults for Delphi Diagnostics.
References
2. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013 Aug 22;500(7463):415–21.
3. Kucab JE, Zou X, Morganella S, Joel M, Nanda AS, Nagy E, et al. A Compendium of Mutational Signatures of Environmental Agents. Cell. 2019 May 2;177(4):821–836.e16.
4. Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature. 2020 Feb;578(7793):94–101.
5. Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012 May 25;149(5):979–93.
6. Alexandrov LB, Ju YS, Haase K, Van Loo P, Martincorena I, Nik-Zainal S, et al. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016 Nov 4;354(6312):618–22.
7. Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017 Apr;23(4):517-25.
8. Brady SW, Gout AM, Zhang J. Therapeutic and prognostic insights from the analysis of cancer mutational signatures. Trends Genet. 2022 Feb 1;38(2):194–208.
9. Islam SA, Díaz-Gay M, Wu Y, Barnes M, Vangara R, Bergstrom EN, et al. Uncovering novel mutational signatures by de novo extraction with SigProfilerExtractor. Cell Genom. 2022 Nov 9;2(11).
10. Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TB, Veeriah S, et al. Tracking the evolution of non–small-cell lung cancer. N Engl J Med. 2017 Jun 1;376(22):2109–21.
11. Samur MK, Aktas Samur A, Fulciniti M, Szalat R, Han T, Shammas M, et al. Genome-wide somatic alterations in multiple myeloma reveal a superior outcome group. J Clin Oncol. 2020 Sep 20;38(27):3107–18.
12. Greenman C, Wooster R, Futreal PA, Stratton MR, Easton DF. Statistical analysis of pathogenicity of somatic mutations in cancer. Genetics. 2006 Aug 1;173(4):2187–98.
13. Parmigiani G, Boca S, Lin J, Kinzler KW, Velculescu V, Vogelstein B. Design and analysis issues in genome-wide somatic mutation studies of cancer. Genomics. 2009 Jan 1;93(1):17–21.
14. Lee DD, Seung HS. Learning the parts of objects by non-negative matrix factorization. nature. 1999 Oct 21;401(6755):788–91.
15. Tan VY, Févotte C. Automatic relevance determination in nonnegative matrix factorization with the β-divergence. IEEE Trans Pattern Anal Mach Intell. 2013 Jul;35(7):1592–605.
16. Rosales RA, Drummond RD, Valieris R, Dias-Neto E, da Silva IT. signeR: an empirical Bayesian approach to mutational signature discovery. Bioinformatics. 2017 Jan 1;33(1):8–16.
17. Grabski IN, Trippa L, Parmigiani G. Bayesian multi-study non-negative matrix factorization for mutational signatures. Genome Biol.2025 Apr 16;26(1):98.
18. Medo M, Ng CKY, Medová M. A comprehensive comparison of tools for fitting mutational signatures. Nat Commun. 2024 Nov 2;15(1):9467.
19. Fischer A, Illingworth CJ, Campbell PJ, Mustonen V. EMu: probabilistic inference of mutational processes and their localization in the cancer genome. Genome Biol. 2013 Apr 29;14(4):R39.
20. Shiraishi Y, Tremmel G, Miyano S, Stephens M. A simple model-based approach to inferring and visualizing cancer mutation signatures. PLoS Genet. 2015 Dec 2;11(12):e1005657.
21. K. Gori and A. Baez-Ortega. sigfit: flexible Bayesian inference of mutational signatures. BioRxiv. 2020 Jan 17:372896.
22. Liu M, Wu Y, Jiang N, Boot A, Rozen SG. mSigHdp: hierarchical Dirichlet process mixture modeling for mutational signature discovery. NAR Genom Bioinform. 2023 Mar 1;5(1):lqad005.
23. Zito A, Miller JW. Compressive Bayesian non-negative matrix factorization for mutational signatures analysis. arXiv preprint arXiv:2404.10974. 2024 Apr 17.
24. Hansen B, Grabski IN, Parmigiani G, De Vito R. Bayesian Probit Multi-Study Non-negative Matrix Factorization for Mutational Signatures. arXiv preprint arXiv:2502.01468. 2025 Feb 3.
25. Taylor-Weiner A, Aguet F, Haradhvala NJ, Gosai S, Anand S, Kim J, et al. Scaling computational genomics to millions of individuals with GPUs. Genome Biol. 2019 Nov 1;20(1):228.
26. Li S, Crawford FW, Gerstein MB. Using sigLASSO to optimize cancer mutation signatures jointly with sampling likelihood. Nat Commun. 2020 Jul 17;11(1):3575.
27. Lal A, Liu K, Tibshirani R, Sidow A, Ramazzotti D. De novo mutational signature discovery in tumor genomes using SparseSignatures. PLoS Comput Biol. 2021 Jun 28;17(6):e1009119.
28. Jin H, Gulhan DC, Geiger B, Ben-Isvy D, Geng D, et al. Accurate and sensitive mutational signature analysis with MuSiCal. Nat Genet. 2024 Mar;56(3):541–52.
29. Xue C, Zito A, Miller JW. Improved control of Dirichlet location and scale near the boundary. arXiv preprint arXiv:2410.13050. 2024 Oct 16.
30. Landy JM, Basava N, Parmigiani G. bayesNMF: Fast Bayesian Poisson NMF with Automatically Learned Rank Applied to Mutational Signatures. arXiv preprint arXiv:2502.18674. 2025 Feb 25.
31. Pandey P, Arora S, Rosen GL. MetaMutationalSigs: comparison of mutational signature refitting results made easy. Bioinformatics. 2022 Apr 15;38(8):2344–47.
32. Vöhringer H, Hoeck AV, Cuppen E, Gerstung M. Learning mutational signatures and their multidimensional genomic properties with TensorSignatures. Nat Commun. 2021 Jun 15;12(1):3628.
33. Robinson W, Sharan R, Leiserson MD. Modeling clinical and molecular covariates of mutational process activity in cancer. Bioinformatics. 2019 Jul;35(14):i492–500.
34. Otlu B, Alexandrov LB. Evaluating topography of mutational signatures with SigProfilerTopography. Genome Biol. 2025 May 20;26(1):134.
35. Lang I, Landy J, Parmigiani G. Bayesian Non-Negative Matrix Factorization with Correlated Mutation Type Probabilities for Mutational Signatures. ArXiv [Preprint]. 2025 Jun 28:arXiv:2506.15855v2.
36. Pancotti C, Rollo C, Codice F, Birolo G, Fariselli P, Sanavia T. MUSE-XAE: MUtational Signature Extraction with eXplainable AutoEncoder enhances tumour types classification. Bioinformatics. 2024 May 1;40(5):btae320.
37. Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019 Jan 8;47(D1):D941-D947.
38. Gehring JS, Fischer B, Lawrence M, Huber W. SomaticSignatures: inferring mutational signatures from single-nucleotide variants. Bioinformatics. 2015 Nov 15;31(22):3673–5.
39. Ardin M, Cahais V, Castells X, Bouaoun L, Byrnes G, Herceg Z, et al. MutSpec: a Galaxy toolbox for streamlined analyses of somatic mutation spectra in human and mouse cancer genomes. BMC bioinformatics. 2016 Apr 18;17(1):170.
40. Huang PJ, Chiu LY, Lee CC, Yeh YM, Huang KY, Chiu CH, et al. mSignatureDB: a database for deciphering mutational signatures in human cancers. Nucleic acids research. 2018 Jan 4;46(D1):D964–70.
41. Díaz-Gay M, Vila-Casadesús M, Franch-Expósito S, Hernández-Illán E, Lozano JJ, Castellví-Bel S. Mutational Signatures in Cancer (MuSiCa): a web application to implement mutational signatures analysis in cancer samples. BMC bioinformatics. 2018 Jun 14;19(1):224.
42. Degasperi A, Amarante TD, Czarnecki J, Shooter S, Zou X, Glodzik D, et al. A practical framework and online tool for mutational signature analyses show intertissue variation and driver dependencies. Nature cancer. 2020 Feb 14;1(2):249–63.
43. Wang S, Tao Z, Wu T, Liu XS. Sigflow: an automated and comprehensive pipeline for cancer genome mutational signature analysis. Bioinformatics. 2021 Jul 12;37(11):1590-1592.
44. Fantini D, Meeks JJ. Analysis of Mutational Signatures Using the mutSignatures R Library. Methods Mol Biol. 2023;2684:45-57.
45. Rustad EH, Nadeu F, Angelopoulos N, Ziccheddu B, Bolli N, Puente XS, et al. mmsig: a fitting approach to accurately identify somatic mutational signatures in hematological malignancies. Commun Biol. 2021 Mar 29;4(1):424.
46. Chevalier A, Yang S, Khurshid Z, Sahelijo N, Tong T, Huggins JH, et al. The mutational signature comprehensive analysis toolkit (musicatk) for the discovery, prediction, and exploration of mutational signatures. Cancer Res. 2021 Dec 1;81(23):5813–7.
47. Manders F, Brandsma AM, de Kanter J, Verheul M, Oka R, van Roosmalen MJ, et al. MutationalPatterns: the one stop shop for the analysis of mutational processes. Bmc Genomics. 2022 Feb 15;23(1):134.
48. Díaz-Gay M, Vangara R, Barnes M, Wang X, Islam SMA, Vermes I, et al. Assigning mutational signatures to individual samples and individual somatic mutations with SigProfilerAssignment. bioRxiv [Preprint]. 2023 Jul 11:2023.07.10.548264
49. Steyaert S, Pizurica M, Nagaraj D, Khandelwal P, Hernandez-Boussard T, Gentles AJ, et al. Multimodal data fusion for cancer biomarker discovery with deep learning. Nat Mach Intell. 2023 Apr;5(4):351–62.
50. Cheerla A, Gevaert O. Deep learning with multimodal representation for pancancer prognosis prediction. Bioinformatics. 2019 Jul;35(14):i446–54.
51. Argelaguet R, Velten B, Arnol D, Dietrich S, Zenz T, Marioni JC, et al. Multi‐Omics Factor Analysis—a framework for unsupervised integration of multi‐omics data sets. Mol Syst Biol. 2018 Jun;14(6):e8124.
52. Singh A, Shannon CP, Gautier B, Rohart F, Vacher M, Tebbutt SJ, et al. DIABLO: an integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics. 2019 Sep 1;35(17):3055–62.
53. Afsari B, Kuo A, Zhang Y, Li L, Lahouel K, Danilova L, et al. Supervised mutational signatures for obesity and other tissue-specific etiological factors in cancer. Elife. 2021 Jan 25;10:e61082.
54. Park JE, Smith MA, Van Alsten SC, Walens A, Wu D, Hoadley KA, Troester MA, Love MI. Diffsig: Associating Risk Factors With Mutational Signatures. bioRxiv [Preprint]. 2023 Feb 10:2023.02.09.527740. Update in: Cancer Epidemiol Biomarkers Prev. 2024 May 1;33(5):721-730.
55. Mealey NE, O'Sullivan DE, Pader J, Ruan Y, Wang E, Quan ML, et al. Mutational landscape differences between young-onset and older-onset breast cancer patients. BMC Cancer. 2020 Mar 12;20(1):212.