Commentary
Uveal melanoma (UM) is a rare cancer that affects the choroid and, less frequently, the ciliary body or the iris (for recent reviews see [1-3]). Despite a profound knowledge of the oncogenic mechanisms behind UM tumorigenesis and despite an accurate cytogenetic and molecular prognosis, only limited advances have been made in UM therapy. Therapies targeting mitogen-activated protein (MAP)-kinases have largely failed [4] and immune checkpoint blockers have met with limited success [5,6]. The latter is likely explained by the extremely low mutational burden of 0.5 - 1.1 mutations per megabase [7,8] which translates to 17 [8] to 30 [9] non-synonymous mutations in protein coding sequences per exome and therefore to the generation of few immunogenic neo-antigens. The lack of response to therapies that target downstream effectors of the oncogenic mutations is probably due to the concomitant GNAQ/11 dependent activation of the yes associated protein 1 gene, YAP1 [10,11] that is not inhibited by MAP-kinase inhibitors.
Uveal and cutaneous melanoma are both generated through the transformation of neural crest-derived melanocytes, yet they show different mutations and chromosomal aberrations that drive oncogenesis, a very different mutational burden, low in uveal and high in cutaneous melanoma [12], and different mutational signatures indicating a different etiology [13].
UM is driven by hot spot oncogenic mutations that affect the two G-protein α-subunit Q and 11 genes (GNAQ [14], GNA11 [15]) or, much less frequently [16], the G-protein coupled receptor cysteinyl leukotriene receptor 2 gene (CYSLTR2 [17]) or the phospholipase C beta 4 gene (PLCB4) [7]. Approximately one third of the tumors also carry mutations in the BRCA1-associated protein 1 gene (BAP1) [16], a tumor suppressor gene whose function is depleted through mutation of one and loss of the other allele [18]. Alternatively, hotspot mutation in the splicing factor 3b subunit 1 gene (SF3B1) [19,20] or the serine and arginine rich splicing factor 2 (SRSF2) gene [9,21] likely affect splicing of various genes, thereby creating oncogenic splice variants [22-24]. Interestingly, the splicing factor mutations in UM are different from those observed in the same genes in blood cancers [25,26]. BAP1 is associated with a high [18] and SF3B1 with an intermediate metastatic risk [27]. Cases without any mutation of these two genes have a low metastatic risk. Further frequent gain-of-function mutations in the 5’ part of the coding sequence of the eukaryotic translation initiation factor 1A X-linked gene (EIF1AX) are associated with a low risk of metastasis [20,28]. These mutations likely support a specific tumor development path that does not lead to a metastatic potential.
Chromosomal copy number alterations, mainly monosomy of chromosome 3 and amplification of chromosome 8q, are hallmarks of UM at high risk of metastasis. The analysis of the mutations, copy number alterations, DNA-methylation and RNA expression performed by The Cancer Genome Atlas consortium allows for the identification of two subtypes with different metastatic risks that are characterized by profound differences at all levels of molecular characterization [8]. Each subtype can further be subdivided albeit with less robust discriminators [29-31,8]. The molecular classification is confirmed by data fusion techniques that have been developed for the integration of multi-domain molecular data [32].
GNAQ, GNA11, CYSLTR2 mutations are, with very few exceptions, mutually exclusive [16,9] and are likely to be cancer-initiating mutations. The metastasis drivers BAP1 and SF3B1 are also mutually exclusive with, however, more frequent exceptions [9,16]. PLCB4 mutations have also been described to occur in a mutually-exclusive manner with the other initiating mutations yet the isoforms PLCB1 and PLCB2 have been found in GNAQ-mutated cases [13]. As a consequence, low risk UM carry a single recurrent mutation (GNAQ, GNA11, CYSLTR2 or PLCB4), with in some cases, in addition a mutation in EIF1AX, and high risk cases carry two recurrent mutations, one of the former four and either BAP1 or SF3B1.
The combination of a hotspot mutation in a G-protein α-subunit and a protein-truncating mutation in a tumor suppressor gene would thus be sufficient to form a highly aggressive metastasized tumor that is resistant to chemotherapy, targeted therapy and immune checkpoint blockers and causes the death of the patient within a year of diagnosis of metastases. In favor of this hypothesis comes a genetically engineered mouse model, where the expression of a transgenic GNAQ gene carrying the Q209L mutation is driven by a melanocyte inducing transcription factor, MITF, responsive promoter. In these animals, the single mutation is apparently sufficient to drive uveal melanomagenesis with high penetrance and short latency despite low expression levels of mutated GNAQ [33]. 94% of these animals even presented lung metastases [33]. However, when the expression of mutated GNAQ is induced in adult animals it is not sufficient to generate uveal melanomas [33] indicating that additional events are required.
We envisage five possible scenarios: i) at present, there are data on sequenced exomes from 139 UM [9] and sequencing many more cases might reveal other recurrent mutations that contribute to tumorigenesis and/or metastasization; ii) there might be frequent mutations that are not evidenced by exome sequencing, such as mutations in regulatory elements or non-coding RNAs; iii) copy number alterations, especially those affecting so far unknown elements on chromosome 3 in addition to BAP1 mutations might be necessary; iv) several of the genes that showed mutations in only a few or even a single case might act as secondary drivers; v) a combination of the above.
Scenario #1 can be tested by continuing to sequence the exomes of UM. Scenario #2 can be tested by whole genome sequencing; yet, the first paper on whole genome sequencing did not report any specific non-coding mutations [34]. Scenario #3 offers some obvious clues given the high frequency and the high impact of monosomy of chromosome 3, yet the molecular players in addition to BAP1 have not been identified and, given the considerable effort dedicated to this aspect, are apparently difficult to identify. The sequencing studies would anyway indicate that the missing actor on chromosome 3 is not linked to a somatic mutation but to gene dosage effects. The MYC Proto-Oncogene has been thought to account for the dismal effects of chromosome 8q amplification; yet, there is no direct evidence for this claim, which has been challenged by a study indicating the ArfGAP With SH3 Domain, Ankyrin Repeat And PH Domain 1 gene, ASAP1 (also named DDEF), plays this role [35]. In fact, in our merged gene expression dataset [13], ASAP1 but not MYC is significantly associated with chr8q gain and BAP1 mutation. Given the scattered distribution, even ASAP1 is unlikely to explain all of the chr8q amplification effect (Figure 1).

Figure 1: Expression analysis of MYC and ASAP1. Gene expression data were collected from three publicly available cohorts of primary UM cases [45-48,8] and merged as previously described [13]. The cases for which copy number alteration data and somatic mutation data were available were interrogated for the expression of MYC and ASAP1. A – MYC and ASAP1 expression in cases with and without amplification of chr8q, B – MYC and ASAP1 expression in cases with and without BAP1 mutations. Gene expression data are indicated as arbitrary intensity units, *** = p<0.001; the horizontal line indicates the mean value.
We addressed the fourth scenario by analyzing all the mutations identified by exome sequencing in 139 UM (Figure 2). Our data show that secondary mutations are significantly enriched in the calcium signaling and other pathways in which also the four initiating mutations (GNAQ, GNA11, CYSLTR2 and PLCB4) are annotated. Almost all nodes of the Kyoto Encyclopedia of Genes and Genomes (KEGG) calcium signaling pathway (map04020) contain at least one gene that is mutated in at least one of the 139 UM. This is highly significant (adjusted p<0.0004) despite the fact that gene annotations are extremely incomplete and biased towards genes for which there is more information available. A similar analysis done by a literature search and homology analyses identifies many more genes that are likely to act in the same pathway as the initiating mutations (our unpublished observation). The analysis of potential secondary drivers also led to the identification of several oncogenic hot spot mutations and heterozygous tumor suppressor gene-truncating mutations [13]. The expression values of many of the genes carrying secondary mutations were significantly associated with disease free survival [13].
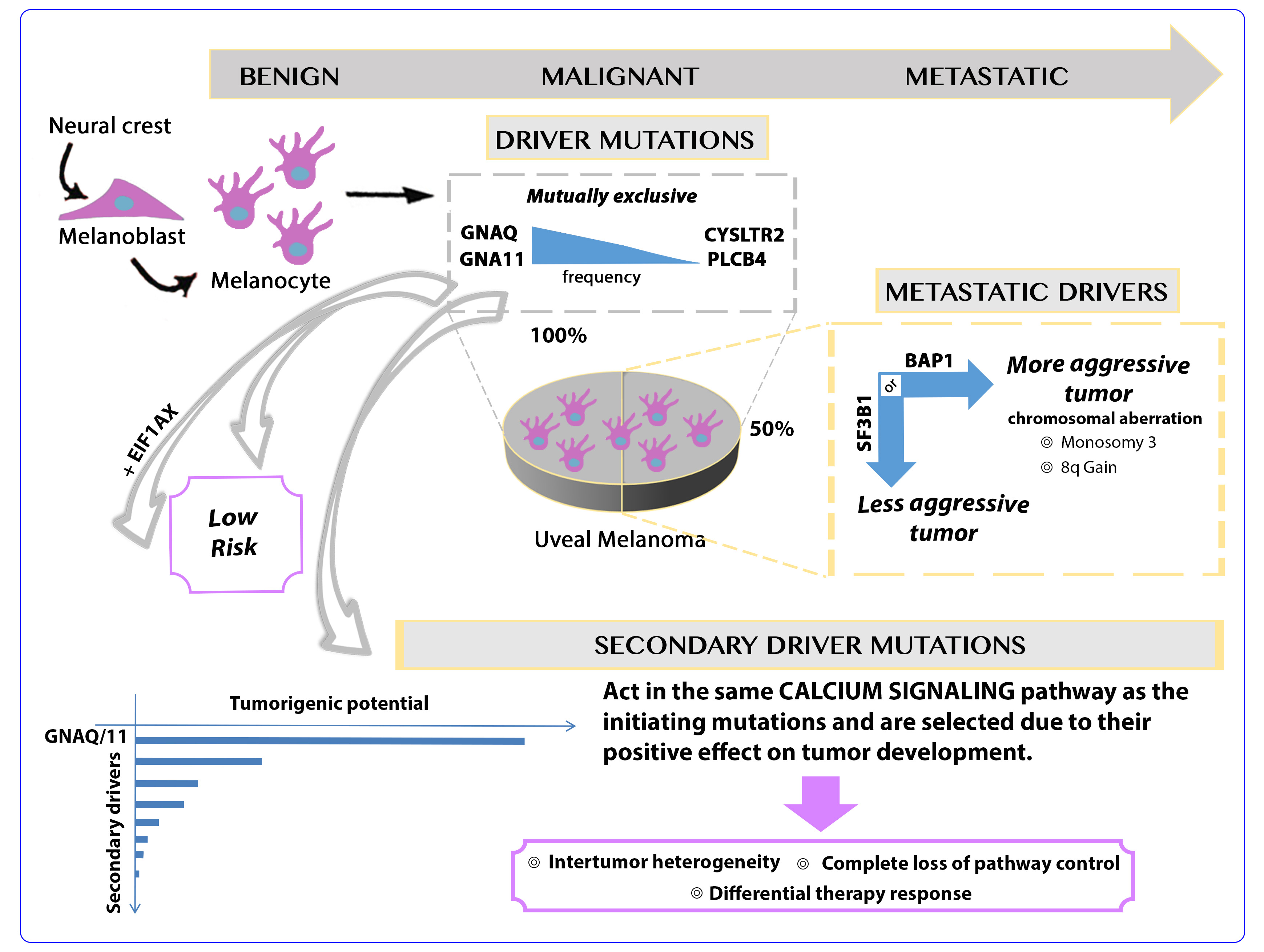
Figure 2: Graphical abstract of the concept of secondary drivers.
Unfortunately, we did not have access to the DNA of the cases analyzed by exome sequencing and were therefore unable to validate these mutations by Sanger sequencing. We followed up one of these mutations, in the protein tyrosine kinase 2β gene (PTK2B). In addition to the two mutations identified by exome sequencing, we could identify two other ones. Two mutations are in the kinase domain and the other two are in the focal-adhesion-targeting domain; yet, a potential oncogenic effect of these mutations is not evident [13].
These observations lead to important conclusions: i) our present distinction of driver and passenger mutations is probably too simple. In addition to strong primary drivers there are many other genes carrying mutations that can affect gene function. If this effect is negative, these mutations are selected against, if the effect is neutral these mutations are carried on as passenger mutations, and if the effect is positive they will be selected. Genetic germline variants influence cancer risk, with very few variants showing a strong effect [36] and many others having very limited yet measurable effects [37]. If this is translated to somatic mutations, we would normally expect very few primary drivers with a strong effect on tumorigenesis followed by secondary drivers that are selected due to their variably positive effect on tumor development; ii) secondary drivers generate inter tumor heterogeneity and might determine the complex therapy responses observed in the clinics since each tumor contains different secondary drivers in addition to a common primary driver; iii) in UM, secondary drivers appear to occur in the same pathway as the primary drivers, in the calcium-signaling pathway (Figure 2). If confirmed, this could indicate that a single mutation is not sufficient to entirely derange an important intracellular pathway, only a second hit in the same pathway determines complete loss of control. This makes sense biologically since the hypothesis that a single mutation is sufficient, likely determines an exaggerated cancer risk, not observed in the real world.
The hypothesis of secondary driver mutations in the same pathway as the primary driver is sustained by a recent analysis of 35 metastatic UM by sequencing a panel of 500 genes commonly involved in cancer. In this study, several additional mutations in genes encoding for factors acting in the G-protein signaling pathway were identified although the analysis of only 35 cases did not allow for enrichment analyses and the limited complexity of the gene panel might have missed many mutations [38].
We also addressed UM mutations in a more general manner by analyzing mutational signatures. Uveal and cutaneous melanoma both show a preponderance of C>T transitions yet they occur in different sequence contexts. Two methods to classify mutation patterns have been developed, and considering the actually mutated nucleotide and one or two others, yield 14 [39] or 96 [40] possibilities. Since each tumor shows more than one of these possibilities, they must be collapsed into a signature. The algorithms used to do this are to some extent arbitrary and can be heuristically considered for the potential to identify signatures linked to specific tumors and/or specific etiological factors [40]. Alexandrov’s algorithm correctly identifies a signature associated to exposure to ultraviolet light that is active in cutaneous [41] but not uveal [8] melanoma indicating that the latter is not caused by UV light, which in fact is absorbed by the vitreous body [42]. However, Alexandrov’s algorithm does not identify any signature that is clearly active in UM [8]. We therefore applied an alternative approach to the calculation of mutational signatures based on sparse dictionary learning [43]. Two of these signatures capture more of the mutational spectrum of UM than Alexandrov’s signatures do, due to the fact that the main consensus of NCG (where C is the mutated nucleotide, N= any nucleotide) was expanded to NCG and CCN. The two UM signatures are not active in cutaneous melanoma, further sustaining the hypothesis of different etiological factors being at work (Figure 3).

Figure 3: Mutational signatures. Mutational signatures were calculated applying sparse dictionary learning [43] for uveal (A) and cutaneous (B) melanoma considering the actually mutated, the preceding and the following nucleotides. For each melanoma type the part of the most informative signature that shows the frequency of the mutated triplets with a central (mutated) C is shown.
If the double hit in the oncogenic pathway can be confirmed for UM there would be no reason to assume that other cancers behave differently. The extraordinarily low mutational burden of UM makes this analysis easier, yet it should be possible to see the same mechanism at work in other cancers. But does this affect therapy? Mechanisms of resistance to targeted therapy are manifold [44]. Activation of upstream or downstream-signaling nodes and parallel-signaling pathways to activate a common downstream pathway are among these mechanisms [44] and are prone to secondary driver mutations. As a consequence, we are proposing to continue to sequence the exomes of primary and metastatic UM since a large case collection will eventually highlight secondary drivers of low frequency that will allow for a fine dissection of the oncogenic pathway driven by GNAQ and GNA11. Future clinical trials with targeted therapy should comprise exome sequencing in order to allow for a correlation between rare responses and specific mutational patterns.
References
2. Zeschnigk M, Lohmann DR. Prognostic testing in uveal melanoma. In: Pfeffer U, editor. Cancer genomics: molecular classification, prognosis and response prediction. Springer Science & Business Media; 2013 Feb 12.
3. Amaro A, Gangemi R, Piaggio F, Angelini G, Barisione G, Ferrini S, Pfeffer U. The biology of uveal melanoma. Cancer and Metastasis Reviews. 2017 Mar 1;36(1):109-40.
4. Croce M, Ferrini S, Pfeffer U, Gangemi R. Targeted therapy of uveal melanoma: Recent failures and new perspectives. Cancers. 2019 Jun;11(6):846.
5. Fountain E, Bassett RL, Cain S, Posada L, Gombos DS, Hwu P, Bedikian A, Patel SP. Adjuvant Ipilimumab in High-Risk Uveal Melanoma. Cancers. 2019 Feb;11(2):152.
6. Bol KF, Ellebaek E, Hoejberg L, Bagger MM, Larsen MS, Klausen TW, Køhler UH, Schmidt H, Bastholt L, Kiilgaard JF, Donia M. Real-world impact of immune checkpoint inhibitors in metastatic uveal melanoma. Cancers. 2019 Oct;11(10):1489
7. Johansson P, Aoude LG, Wadt K, Glasson WJ, Warrier SK, Hewitt AW, Kiilgaard JF, Heegaard S, Isaacs T, Franchina M, Ingvar C. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget. 2016 Jan 26;7(4):4624.
8. Robertson AG, Shih J, Yau C, Gibb EA, Oba J, Mungall KL, Hess JM, Uzunangelov V, Walter V, Danilova L, Lichtenberg TM. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer cell. 2017 Aug 14;32(2):204-20.
9. Field MG, Durante MA, Anbunathan H, Cai LZ, Decatur CL, Bowcock AM, Kurtenbach S, Harbour JW. Punctuated evolution of canonical genomic aberrations in uveal melanoma. Nature communications. 2018 Jan 9;9(1):116.
10. Yu FX, Luo J, Mo JS, Liu G, Kim YC, Meng Z, Zhao L, Peyman G, Ouyang H, Jiang W, Zhao J. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer cell. 2014 Jun 16;25(6):822-30.
11. Feng X, Degese MS, Iglesias-Bartolome R, Vaque JP, Molinolo AA, Rodrigues M, Zaidi MR, Ksander BR, Merlino G, Sodhi A, Chen Q. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer cell. 2014 Jun 16;25(6):831-45.
12. van der Kooij MK, Speetjens FM, van der Burg SH, Kapiteijn E. Uveal Versus Cutaneous Melanoma; Same Origin, Very Distinct Tumor Types. Cancers. 2019 Jun;11(6):845.
13. Piaggio F, Tozzo V, Bernardi C, Croce M, Puzone R, Viaggi S, Patrone S, Barla A, Coviello D, J Jager M, Van Der Velden PA. Secondary Somatic Mutations in G-Protein-Related Pathways and Mutation Signatures in Uveal Melanoma. Cancers. 2019 Nov;11(11):1688.
14. Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, Simpson EM, Barsh GS, Bastian BC. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009 Jan;457(7229):599.
15. Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, Sozen MM. Mutations in GNA11 in uveal melanoma. New England Journal of Medicine. 2010 Dec 2;363(23):2191-9.
16. Dono M, Angelini G, Cecconi M, Amaro A, Esposito AI, Mirisola V, Maric I, Lanza F, Nasciuti F, Viaggi S, Gualco M. Mutation frequencies of GNAQ, GNA11, BAP1, SF3B1, EIF1AX and TERT in uveal melanoma: detection of an activating mutation in the TERT gene promoter in a single case of uveal melanoma. British journal of cancer. 2014 Feb;110(4):1058.
17. Moore AR, Ceraudo E, Sher JJ, Guan Y, Shoushtari AN, Chang MT, Zhang JQ, Walczak EG, Kazmi MA, Taylor BS, Huber T. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nature genetics. 2016 Jun;48(6):675.
18. Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, Council ML, Matatall KA, Helms C, Bowcock AM. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010 Dec 3;330(6009):1410-3.
19. Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM (2013) Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nature genetics 45 (2):133-135. doi:10.1038/ng.2523
20. Martin M, Maßhöfer L, Temming P, Rahmann S, Metz C, Bornfeld N, van de Nes J, Klein-Hitpass L, Hinnebusch AG, Horsthemke B, Lohmann DR. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nature genetics. 2013 Aug;45(8):933.
21. Park JJ, Diefenbach RJ, Joshua AM, Kefford RF, Carlino MS, Rizos H. Oncogenic signaling in uveal melanoma. Pigment cell & melanoma research. 2018 Nov;31(6):661-72.
22. Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, Corson L. Cancer-associated SF3B1 hotspot mutations induce cryptic 3′ splice site selection through use of a different branch point. Cell reports. 2015 Nov 3;13(5):1033-45.
23. DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, Jamieson CH, Carson D, Kipps TJ, Frazer KA. Transcriptome sequencing reveals potential mechanism of cryptic 3’splice site selection in SF3B1-mutated cancers. PLoS computational biology. 2015 Mar 13;11(3):e1004105.
24. Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, Tirode F, Constantinou A, Piperno-Neumann S, Roman-Roman S, Dutertre M. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nature communications. 2016 Feb 4;7:10615.
25. van Poppelen NM, Drabarek W, Smit KN, Vaarwater J, Brands T, Paridaens D, Kilic E, de Klein A (2019) SRSF2 Mutations in Uveal Melanoma: A Preference for In-Frame Deletions? Cancers 11 (8). doi:10.3390/cancers11081200
26. Quesada V, Conde L, Villamor N, Ordóñez GR, Jares P, Bassaganyas L, Ramsay AJ, Beà S, Pinyol M, Martínez-Trillos A, López-Guerra M. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nature genetics. 2012 Jan;44(1):47.
27. Yavuzyigitoglu S, Koopmans AE, Verdijk RM, Vaarwater J, Eussen B, Van Bodegom A, Paridaens D, Kiliç E, de Klein A, Rotterdam Ocular Melanoma Study Group. Uveal melanomas with SF3B1 mutations: a distinct subclass associated with late-onset metastases. Ophthalmology. 2016 May 1;123(5):1118-28.
28. Ewens KG, Kanetsky PA, Richards-Yutz J, Purrazzella J, Shields CL, Ganguly T, Ganguly A. Chromosome 3 status combined with BAP1 and EIF1AX mutation profiles are associated with metastasis in uveal melanoma. Investigative ophthalmology & visual science. 2014 Aug 1;55(8):5160-7.
29. Vichitvejpaisal P, Dalvin LA, Mazloumi M, Ewens KG, Ganguly A, Shields CL (2019) Genetic Analysis of Uveal Melanoma in 658 Patients Using the Cancer Genome Atlas Classification of Uveal Melanoma as A, B, C, and D. Ophthalmology 126 (10):1445-1453. doi:10.1016/j.ophtha.2019.04.027
30. Jager MJ, Brouwer NJ, Esmaeli B. The Cancer Genome Atlas Project: an integrated molecular view of uveal melanoma. Ophthalmology. 2018 Aug 1;125(8):1139-42.
31. Bakhoum MF, Esmaeli B. Molecular Characteristics of Uveal Melanoma: Insights from the Cancer Genome Atlas (TCGA) Project. Cancers. 2019 Aug;11(8):1061.
32. Pfeffer M, Uschmajew A, Amaro A, Pfeffer U. Data fusion techniques for the integration of multi-domain genomic data from uveal melanoma. Cancers. 2019 Oct;11(10):1434.
33. Huang JL, Urtatiz O, Van Raamsdonk CD. Oncogenic G protein GNAQ induces uveal melanoma and intravasation in mice. Cancer research. 2015 Aug 15;75(16):3384-97.
34. Royer-Bertrand B, Torsello M, Rimoldi D, El Zaoui I, Cisarova K, Pescini-Gobert R, Raynaud F, Zografos L, Schalenbourg A, Speiser D, Nicolas M. Comprehensive genetic landscape of uveal melanoma by whole-genome sequencing. The American Journal of Human Genetics. 2016 Nov 3;99(5):1190-8.
35. Ehlers JP, Worley L, Onken MD, Harbour JW. DDEF1 is located in an amplified region of chromosome 8q and is overexpressed in uveal melanoma. Clinical Cancer Research. 2005 May 15;11(10):3609-13.
36. Walsh T, Casadei S, Coats KH, Swisher E, Stray SM, Higgins J, Roach KC, Mandell J, Lee MK, Ciernikova S, Foretova L. Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. Jama. 2006 Mar 22;295(12):1379-88.
37. Zhang Y, Wilcox AN, Zhang H, Choudhury PP, Easton DF, Milne RL, Simard J, Hall P, Michailidou K, Dennis J, Schmidt MK. Assessment of Polygenic Architecture and Risk Prediction based on Common Variants Across Fourteen Cancers. bioRxiv. 2019 Jan 1:723825.
38. Shain AH, Bagger MM, Yu R, Chang D, Liu S, Vemula S, Weier JF, Wadt K, Heegaard S, Bastian BC, Kiilgaard JF. The genetic evolution of metastatic uveal melanoma. Nature genetics. 2019 Jul;51(7):1123-30.
39. Shiraishi Y, Tremmel G, Miyano S, Stephens M. A simple model-based approach to inferring and visualizing cancer mutation signatures. PLoS genetics. 2015 Dec 2;11(12):e1005657.
40. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S. Signatures of mutational processes in human cancer. Nature. 2013 Aug;500(7463):415.
41. Akbani R, Akdemir KC, Aksoy BA, Albert M, Ally A, Amin SB, Arachchi H, Arora A, Auman JT, Ayala B, Baboud J. Genomic classification of cutaneous melanoma. Cell. 2015 Jun 18;161(7):1681-96.
42. Balazs EA. Studies on the Structure of the Vitreous Body*: I. The Absorption of Ultraviolet Light. American journal of ophthalmology. 1954 Jul 1;38(1):21-8.
43. Tozzo V, Barla A. Cancer Mutational Signatures Identification with Sparse Dictionary Learning. In: Cham. Computational Intelligence Methods for Bioinformatics and Biostatistics. Springer International Publishing, 2019; pp 32-41
44. Sabnis AJ, Bivona TG. Principles of resistance to targeted cancer therapy: lessons from basic and translational cancer biology. Trends in molecular medicine. 2019 Jan 24.
45. Herlihy N, Dogrusöz M, Van Essen TH, Harbour JW, Van Der Velden PA, Van Eggermond MC, Haasnoot GW, Van Den Elsen PJ, Jager MJ. Skewed expression of the genes encoding epigenetic modifiers in high-risk uveal melanoma. Investigative ophthalmology & visual science. 2015 Mar 1;56(3):1447-58.
46. Laurent C, Valet F, Planque N, Silveri L, Maacha S, Anezo O, Hupe P, Plancher C, Reyes C, Albaud B, Rapinat A. High PTP4A3 phosphatase expression correlates with metastatic risk in uveal melanoma patients. Cancer research. 2011 Feb 1;71(3):666-74.
47. Amaro A, Mirisola V, Angelini G, Musso A, Tosetti F, Esposito AI, Perri P, Lanza F, Nasciuti F, Mosci C, Puzone R. Evidence of epidermal growth factor receptor expression in uveal melanoma: inhibition of epidermal growth factor-mediated signalling by Gefitinib and Cetuximab triggered antibody-dependent cellular cytotoxicity. European Journal of Cancer. 2013 Oct 1;49(15):3353-65.
48. Laurent C, Valet F, Planque N, Silveri L, Maacha S, Anezo O, Hupe P, Plancher C, Reyes C, Albaud B, Rapinat A. High PTP4A3 phosphatase expression correlates with metastatic risk in uveal melanoma patients. Cancer research. 2011 Feb 1;71(3):666-74.