Keywords
Influenza, Interferons, Streptococcus Pneumoniae, SPn co-infection, IFN-γ
Commentary
Influenza A virus (IAV) and Streptococcus pneumoniae (SPn) are two major respiratory pathogens in humans. IAV infection alone is often self-limited, and SPn colonization can be found in 5-90% of healthy individuals, as normal flora [1,2]. On the other hand, IAV and SPn co-infection represents a major health concern and is known to cause severe morbidity and mortality in influenza pandemics and epidemics [3-6]. The murine models of IAV and bacterial co-infection are well-established and replicate key characteristics of human patients. Multiple mechanisms have been revealed to be involved in co-pathogenesis, including enhanced bacterial colonization and invasion, suppression of antimicrobial immunity, and acute lung injury. It is likely that these pathogenic mechanisms concomitantly or sequentially promote co-infection progression [7]. Here we will discuss some recent evidence on the role and interplay between type I IFNs (IFN-I), i.e., IFN-α/β, and IFN-g in promoting IAV/SPn co-pathogenesis.
Acute respiratory viral infection typically activates IFN responses in the lung. These IFNs are critical regulators of the balance between antiviral immunity and detrimental inflammation [8,9]. Alveolar macrophages are the main producers of IFN-I at the early stage of IAV infection [10,11]. IFN-I signaling not only limits virus replication but also attenuates inflammatory lung damage [12-14]. Nonetheless, T cell recruitment and activation at the later phase (~7 d) is essential to clear IAV infection. Influenza typically activates IFN-g-biased cytokine production by T cells [8]. IFN-γ is not required for viral clearance but limits lung inflammation [15]. Notably, influenza-induced peak susceptibility to SPn infection coincides with T cell recruitment [16]. Furthermore, T cell depletion restores acute bacterial clearance after IAV/SPn co-infection [17], suggesting a key role of T cells in suppression of innate antibacterial immunity [16,18].
Conversely, immune clearance of SPn infection requires phagocytic cells. Resident alveolar macrophages represent a strong first line of innate defense against extracellular bacteria [19,20], explaining the unproblematic SPn carriage in healthy individuals. Neutrophils play a significant role in bacterial killing especially at the later stage of infection. Recent evidence indicates that the antibacterial function of both alveolar macrophages and neutrophils is impaired by IAV-induced IFN responses, particularly IFN-γ and IFN-I, [21-27], thereby predisposing hosts to secondary bacterial pneumonia.
Compelling evidence from animal models indicates that IAV infection induces alveolar macrophage dysfunction and depletion [16,28-31]. Specifically, we have shown that T cell-derived IFN-γ inhibits alveolar macrophage-mediated bacterial killing during recovery from IAV infection [16]. In agreement, a mathematical model predicts that alveolar macrophage dysfunction is primarily responsible for bacterial outgrowth during co-infection [32]. It has also been shown in BALB/c mice that IAV infection depletes alveolar macrophages [28]. However, the effect of IAV infection on alveolar macrophage survival appears to be dependent on viral virulence and mouse genetic strains. As such, alveolar macrophage levels are maintained after a low dose of A/PR/8 (H1N1) infection in C57BL/6 mice and throughout the course of low-virulent X31 (H3N2) infection in BALB/c mice. Conversely, using both IFN-γ gene-deficient mice as well as in vivo neutralizing antibodies, we have recently demonstrated that IFN-γ induces alveolar macrophage depletion during co-infection progression. Collectively, these findings indicate that IFN-γ not only impairs alveolar macrophage antibacterial function but also their viability during the co-pathogenesis process.
On the other hand, IFN-I signaling promotes inflammatory monocyte recruitment after IAV infection. Monocytes are normally protective against SPn infection alone [33], but their inflammatory property is detrimental to antibacterial defense after IAV infection [34-36], by promoting IFN-I [37] and IFN-g-induced lung epithelial damage [36]. Of particular interest, during recovery from IAV infection, these CCR2-recruited monocytes differentiate into alveolar macrophages to confer prolonged antibacterial protection [38]. The mechanism underlying the phenotypic and functional transition of monocytes remains unclear.
Another common observation during IAV and bacterial co-infection is neutrophil dysregulation [39,40]. As the first responders to microbial infections, neutrophils perform a critical role in host defense against opportunistic pathogens, and their prompt recruitment is crucial to prevent the establishment of infection. In line with that, multiple studies have demonstrated that IAV-induced IFN-I response suppresses neutrophil recruitment and thereby increases host susceptibility to pneumococcal pneumonia [18,22,26,41]. In addition to that, IFN-g signaling also suppresses neutrophil infiltration [17,42]. However, neutrophilic inflammation increases acute lung damage, which can inadvertently exacerbate disease progression [43,44].
Unlike other high-virulent mouse-adapted IAV strains, X31 infection alone does not induce strong IFN-g response. We have recently reported that prior X31 infection induces lethal susceptibility to SPn pneumonia in C57BL/6 but not BALB/c mice. In line with that, X31/SPn co-infection induces prominent IFN-g production in C57BL/6 mice and thereby promotes the hypersusceptibility. Interestingly, the resistant BALB/c mice depend on neutrophils for effective bacterial clearance. In fact, C57BL/6 mice exhibited significantly increased neutrophil infiltration than BALB/c animals, but they were still defective in lung bacterial control. It suggests that IFN-γ may play a role in inhibiting neutrophil function. Indeed, IAV infection has been shown to impair neutrophil antibacterial function [40]. Thus, IFN-γ can play multiple roles in innate suppression during IAV/SPn co-pathogenesis, including impairment of AM function and survival [16,30], inhibition of neutrophil recruitment, and likely neutrophil dysfunction.
During IAV infection alone, it has been shown that rather than regulating IFN-γ production, IFN-I reduces IFN-γR expression and thereby protects Ly6Clo monocytes/macrophages from IFN-γ-induced activation [8]. IFN-I also promotes SPn colonization and transmission following secondary IAV infection [45,46]. Using a similar SPn carriage model, it has been demonstrated that sequential neutralization of IFN-I and IFN-γ pathways provides optimal protection against SPn/IAV co-infection [47]. In agreement with that, we have shown that IFN-I signaling plays a significant role in suppression of acute bacterial clearance. However, IFN-I signaling is essential for preventing IFN-γ hyperproduction and subsequent exacerbation of co-infection. Of note, the susceptibility to secondary bacterial pneumonia peaks around a week after IAV infection, coinciding with T cell IFN-g production [16]. A similar time interval has been observed between the onset of symptoms of viral infection and bacterial pneumonia in humans [48]. Thus, it is likely the IFN-γ plays a dominant role in immune predisposition to SPn superinfection, particularly during recovery from viral infection (Figure 1).
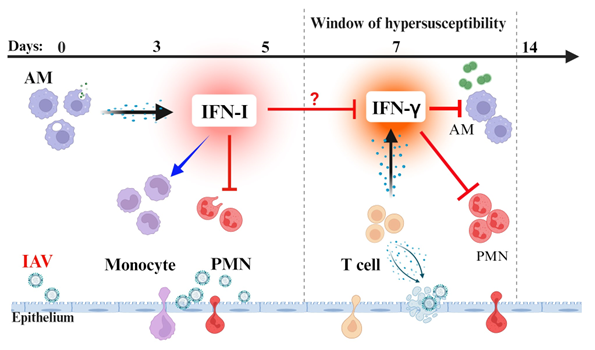
Figure 1: IFN-I and IFN-g in IAV and SPn co-pathogenesis. At the innate phase of IAV infection, alveolar macrophages produce IFN-I to limit viral replication. IFN-I signaling recruits monocytes and limits neutrophil infiltration, resulting in impaired innate defense against SPn infection. At the adaptive phase of influenza, T cells are recruited to clear viruses in the lung. However, T cell-derived IFN-g impairs the antibacterial function of both alveolar macrophages and neutrophils, thereby perpetuating host susceptibility to bacterial pneumonia after influenza. Created with Biorender.com.
In summary, recent evidence indicates that during the early phase of IAV infection, IFN-I plays a key role in suppression of antibacterial immunity by inhibiting neutrophil recruitment; while at the recovery phase of IAV infection, T cell-derived IFN-γ plays a dominant role in suppression of both alveolar macrophages and neutrophils. The sequential IFN-I and IFN-γ signaling results in the prolonged host susceptibility to pneumococcal infection after influenza.
References
2. Rowe HM, Meliopoulos VA, Iverson A, Bomme P, Schultz-Cherry S, Rosch JW. Direct interactions with influenza promote bacterial adherence during respiratory infections. Nat Microbiol. 2019;4(8):1328-36.
3. Metzger DW, Sun K. Immune dysfunction and bacterial coinfections following influenza. Journal of Immunology (Baltimore, Md : 1950). 2013;191(5):2047-52.
4. Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. 2008;198(7):962-70.
5. Gupta RK, George R, Nguyen-Van-Tam JS. Bacterial pneumonia and pandemic influenza planning. Emerg Infect Dis. 2008;14(8):1187-92.
6. Surveillance for pediatric deaths associated with 2009 pandemic influenza A (H1N1) virus infection - United States, April-August 2009. MMWR Morb Mortal Wkly Rep. 2009;58(34):941-7.
7. Melvin JA, Bomberger JM. Compromised Defenses: Exploitation of Epithelial Responses During Viral-Bacterial Co-Infection of the Respiratory Tract. PLoS Pathogens. 2016;12(9):e1005797.
8. Stifter SA, Bhattacharyya N, Pillay R, Florido M, Triccas JA, Britton WJ, et al. Functional Interplay between Type I and II Interferons Is Essential to Limit Influenza A Virus-Induced Tissue Inflammation. PLoS Pathogens. 2016;12(1):e1005378.
9. Major J, Crotta S, Llorian M, McCabe TM, Gad HH, Priestnall SL, et al. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science. 2020;369(6504):712-7.
10. Kumagai Y, Takeuchi O, Kato H, Kumar H, Matsui K, Morii E, et al. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity. 2007;27(2):240-52.
11. Helft J, Manicassamy B, Guermonprez P, Hashimoto D, Silvin A, Agudo J, et al. Cross-presenting CD103+ dendritic cells are protected from influenza virus infection. J Clin Invest. 2012;122(11):4037-47.
12. Arimori Y, Nakamura R, Yamada H, Shibata K, Maeda N, Kase T, et al. Type I interferon limits influenza virus-induced acute lung injury by regulation of excessive inflammation in mice. Antiviral Research. 2013;99(3):230-7.
13. Jiang L, Yao S, Huang S, Wright J, Braciale TJ, Sun J. Type I IFN signaling facilitates the development of IL-10-producing effector CD8+ T cells during murine influenza virus infection. European Journal of Immunology. 2016;46(12):2778-88.
14. Seo SU, Kwon HJ, Ko HJ, Byun YH, Seong BL, Uematsu S, et al. Type I interferon signaling regulates Ly6C(hi) monocytes and neutrophils during acute viral pneumonia in mice. PLoS Pathogens. 2011;7(2):e1001304.
15. Califano D, Furuya Y, Roberts S, Avram D, McKenzie ANJ, Metzger DW. IFN-gamma increases susceptibility to influenza A infection through suppression of group II innate lymphoid cells. Mucosal Immunol. 2018;11(1):209-19.
16. Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat Med. 2008;14(5):558-64.
17. Palani S, Uddin MB, McKelvey M, Shao S, Sun K. Immune predisposition drives susceptibility to pneumococcal pneumonia after mild influenza A virus infection in mice. Front Immunol. 2023;14:1272920.
18. Er JZ, Koean RAG, Ding JL. Loss of T-bet confers survival advantage to Influenza-bacterial Superinfection. EMBO J. 2019;38(1).
19. Sun K, Gan Y, Metzger DW. Analysis of murine genetic predisposition to pneumococcal infection reveals a critical role of alveolar macrophages in maintaining the sterility of the lower respiratory tract. Infection and Immunity. 2011;79(5):1842-7.
20. Yajjala VK, Thomas VC, Bauer C, Scherr TD, Fischer KJ, Fey PD, et al. Resistance to Acute Macrophage Killing Promotes Airway Fitness of Prevalent Community-Acquired Staphylococcus aureus Strains. Journal of Immunology (Baltimore, Md : 1950). 2016;196(10):4196-203.
21. Planet PJ, Parker D, Cohen TS, Smith H, Leon JD, Ryan C, et al. Lambda Interferon Restructures the Nasal Microbiome and Increases Susceptibility to Staphylococcus aureus Superinfection. MBio. 2016;7(1):e01939-15.
22. Cao J, Wang D, Xu F, Gong Y, Wang H, Song Z, et al. Activation of IL-27 signalling promotes development of postinfluenza pneumococcal pneumonia. EMBO Molecular Medicine. 2014;6(1):120-40.
23. Rich HE, McCourt CC, Zheng WQ, McHugh KJ, Robinson KM, Wang J, et al. Interferon Lambda Inhibits Bacterial Uptake during Influenza Superinfection. Infection and Immunity. 2019;87(5).
24. Lee B, Robinson KM, McHugh KJ, Scheller EV, Mandalapu S, Chen C, et al. Influenza-induced type I interferon enhances susceptibility to gram-negative and gram-positive bacterial pneumonia in mice. Am J Physiol Lung Cell Mol Physiol. 2015;309(2):L158-67.
25. Li W, Moltedo B, Moran TM. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of gammadelta T cells. J Virol. 2012;86(22):12304-12.
26. Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest. 2009;119(7):1910-20.
27. Shepardson KM, Larson K, Morton RV, Prigge JR, Schmidt EE, Huber VC, et al. Differential Type I Interferon Signaling Is a Master Regulator of Susceptibility to Postinfluenza Bacterial Superinfection. MBio. 2016;7(3).
28. Ghoneim HE, Thomas PG, McCullers JA. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. Journal of Immunology (Baltimore, Md : 1950). 2013;191(3):1250-9.
29. Shirey KA, Perkins DJ, Lai W, Zhang W, Fernando LR, Gusovsky F, et al. Influenza "Trains" the Host for Enhanced Susceptibility to Secondary Bacterial Infection. mBio. 2019;10(3).
30. Neupane AS, Willson M, Chojnacki AK, Vargas ESCF, Morehouse C, Carestia A, et al. Patrolling Alveolar Macrophages Conceal Bacteria from the Immune System to Maintain Homeostasis. Cell. 2020;183(1):110-25 e11.
31. Verma AK, Bansal S, Bauer C, Muralidharan A, Sun K. Influenza Infection Induces Alveolar Macrophage Dysfunction and Thereby Enables Noninvasive Streptococcus pneumoniae to Cause Deadly Pneumonia. Journal of Immunology (Baltimore, Md : 1950). 2020;205(6):1601-7.
32. Smith AM, Adler FR, Ribeiro RM, Gutenkunst RN, McAuley JL, McCullers JA, et al. Kinetics of coinfection with influenza A virus and Streptococcus pneumoniae. PLoS Pathogens. 2013;9(3):e1003238.
33. Winter C, Herbold W, Maus R, Langer F, Briles DE, Paton JC, et al. Important role for CC chemokine ligand 2-dependent lung mononuclear phagocyte recruitment to inhibit sepsis in mice infected with Streptococcus pneumoniae. Journal of Immunology (Baltimore, Md : 1950). 2009;182(8):4931-7.
34. Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. Journal of Immunology (Baltimore, Md : 1950). 2008;180(4):2562-72.
35. Ellis GT, Davidson S, Crotta S, Branzk N, Papayannopoulos V, Wack A. TRAIL+ monocytes and monocyte-related cells cause lung damage and thereby increase susceptibility to influenza-Streptococcus Pneumoniae Coinfection. EMBO reports. 2015;16(9):1203-18.
36. Schmit T, Guo K, Tripathi JK, Wang Z, McGregor B, Klomp M, et al. Interferon-gamma promotes monocyte-mediated lung injury during influenza infection. Cell Rep. 2022;38(9):110456.
37. Davidson S, Crotta S, McCabe TM, Wack A. Pathogenic potential of interferon alphabeta in acute influenza infection. Nat Commun. 2014;5:3864.
38. Aegerter H, Kulikauskaite J, Crotta S, Patel H, Kelly G, Hessel EM, et al. Influenza-induced monocyte-derived alveolar macrophages confer prolonged antibacterial protection. Nat Immunol. 2020;21(2):145-57.
39. Grunwell JR, Giacalone VD, Stephenson S, Margaroli C, Dobosh BS, Brown MR, et al. Neutrophil Dysfunction in the Airways of Children with Acute Respiratory Failure Due to Lower Respiratory Tract Viral and Bacterial Coinfections. Scientific Reports. 2019;9(1):2874.
40. McNamee LA, Harmsen AG. Both influenza-induced neutrophil dysfunction and neutrophil-independent mechanisms contribute to increased susceptibility to a secondary Streptococcus pneumoniae infection. Infection and Immunity. 2006;74(12):6707-21.
41. Kudva A, Scheller EV, Robinson KM, Crowe CR, Choi SM, Slight SR, et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. Journal of Immunology (Baltimore, Md : 1950). 2011;186(3):1666-74.
42. Palani S, Bansal S, Verma AK, Bauer C, Shao S, Uddin MB, et al. Type I IFN Signaling Is Essential for Preventing IFN-gamma Hyperproduction and Subsequent Deterioration of Antibacterial Immunity during Postinfluenza Pneumococcal Infection. Journal of Immunology (Baltimore, Md : 1950). 2022;209(1):128-35.
43. Aguilera ER, Lenz LL. Inflammation as a Modulator of Host Susceptibility to Pulmonary Influenza, Pneumococcal, and Co-Infections. Front Immunol. 2020;11:105.
44. Castro AM, Cabello-Gutierrez C, Pulido-Camarillo E, Garcia-Garcia AE, Perez-Torres A. Effect of coinfection with influenza virus and bacteria on host damage. Gac Med Mex. 2020;156(4):273-8.
45. Nakamura S, Davis KM, Weiser JN. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J Clin Invest. 2011;121(9):3657-65.
46. Zangari T, Ortigoza MB, Lokken-Toyli KL, Weiser JN. Type I Interferon Signaling Is a Common Factor Driving Streptococcus pneumoniae and Influenza A Virus Shedding and Transmission. mBio. 2021;12(1).
47. Barman TK, Racine R, Bonin JL, Califano D, Salmon SL, Metzger DW. Sequential targeting of interferon pathways for increased host resistance to bacterial superinfection during influenza. PLoS Pathogens. 2021;17(3):e1009405.
48. Brundage JF. Interactions between influenza and bacterial respiratory pathogens: implications for pandemic preparedness. Lancet Infect Dis. 2006;6(5):303-12.