Abstract
Low-frequency somatic mutations accumulate in normal tissues throughout life and contribute to cancer initiation, yet their detection is limited by the sensitivity of conventional sequencing methods. We describe CarcSeq, an error-corrected, targeted next-generation sequencing platform developed to quantify rare cancer driver mutations (CDMs) at variant allele frequencies as low as ~10-4. CarcSeq integrates high-fidelity amplification, unique molecular identifiers, and single-strand consensus sequencing to accurately measure mutant frequency (MF) across a curated panel of sequences encompassing recurrent oncogene and tumor suppressor hotspots. To capture mutation heterogeneity associated with early clonal expansion, the median absolute deviation (MAD) of MF was incorporated as a metric.
Application of CarcSeq to normal and tumor-adjacent human tissues revealed tissue-specific mutational profiles and demonstrated that recurrent driver mutations are detectable in histologically normal samples. Extension to rodent models showed that MAD correlates strongly with strain-, tissue-, and sex-specific spontaneous tumor incidence, supporting its utility as an early biomarker of neoplastic susceptibility. In an exposure study, CarcSeq detected dose- and time-dependent clonal expansion of spontaneous Pik3ca H1047R mutations following administration of the nongenotoxic carcinogen lorcaserin, despite no overall increase in global mutation frequency, highlighting sensitivity to early carcinogenic processes not captured by traditional genotoxicity assays.
Compared with whole-genome, whole-exome, and ultra-deep error-corrected sequencing approaches, CarcSeq balances sensitivity, throughput, and cost by focusing on biologically human cancer-relevant driver mutation hotspots. Together, these findings establish CarcSeq-derived MF and MAD as quantitative, cross-species biomarkers of early clonal expansion with applications in translational carcinogenicity assessment, drug development, and cancer risk modeling.
Keywords
Next-generation sequencing, Cancer, Mutations, CarcSeq, Tumor
Introduction
Over the past decade, genomic evaluation has become central to cancer prevention, early detection of endogenous sources of mutation (e.g. physicochemical processes, free radicals, and enzymatic processes controlling DNA damage and repair), and pharmacogenomic-based therapeutic strategies. Substantial evidence has established the role of lifestyle and environmental determinants in carcinogenesis [1], with cumulative exposures shown to induce somatic mutations despite functional DNA repair mechanisms. Analysis of a cohort comprising 821 non cancer individuals revealed the presence of somatic mutations in histologically normal tissues, occurring at estimated frequencies of 2–6 mutations per million bases [2].
Advanced genomic technologies—including liquid biopsy [3], noninvasive prenatal testing [3], characterization of somatic mosaicism [4], delineation of tumor subclones [5], and lineage tracing [6]—have shown that somatic single-nucleotide variants (SNVs) frequently occur at low variant allele frequencies (VAFs) that evade detection by conventional sequencing approaches. A major challenge in identifying these low-VAF mutations is distinguishing true rare variants from germline polymorphisms, a difficulty compounded by the widespread use of tumor-only sequencing. Although assay and algorithmic improvements have advanced somatic mutation detection, most pipelines are optimized for variants at higher VAFs, which are enriched during clonal expansion. As a result, widely used oncology workflows remain insufficient for reliably detecting very low-frequency events in normal tissues [7], necessitating alternative methodological approaches.
Cancer Driver Mutations as Biomarkers
These considerations underscore the importance of investigating cancer driver mutations (CDMs) as biomarkers for carcinogenicity assessment and for evaluating cancer risks arising from therapeutic, occupational, and environmental exposures [8]. Because CDMs can be quantified directly from DNA (genotypic selection), the analyses can be performed on isolated DNA from any tissue or species [9]. Our initial studies employed an allele-specific amplification method—Allele-specific Competitive Blocker polymerase chain reaction (ACB-PCR)—to quantify 1 mutant allele among 100,000 total alleles (10-5) in DNA samples [9]. While ACB-PCR provided critical insight into CDMs as biomarkers of cancer risk, it was inherently low-throughput, allowing only single-mutation analyses. The advent of next-generation sequencing (NGS) enabled simultaneous interrogation of many CDMs, including rare variants, and spurred development of high-sensitivity approaches [10].
Reconstructing Somatic Evolution
Although the types of mutations present in cancer genomes are well characterized, the timing of their occurrence has remained unclear until recently. Advances in NGS now enable detailed reconstruction of somatic evolution, throughout cellular lifespan, thereby documenting the transition from normal tissue homeostasis to malignant progression [11]. As part of the Pan-Cancer Analysis of Whole Genomes (PCAWG) Consortium of the International Cancer Genome Consortium (ICGC) and The Cancer Genome Atlas (TCGA) [12], whole-genome sequencing of 2,658 cancers identified 47 million point mutations. Additionally, temporal ordering of somatic mutations during tumor evolution was characterized. Among these, 22% were classified as early clonal, 7% as late clonal, 53% as unspecified clonal, and 17% as subclonal [11]. In a focused analysis of 453 known cancer driver genes, 5,913 oncogenic point mutations were detected, of which 29% were early clonal, 5% late clonal, 56% unspecified clonal, and 8% subclonal. These findings demonstrate that cancer driver mutations predominantly arise during early tumor development, contributing to cancer initiation, whereas later stages (late clonal to subclonal) are characterized by the accumulation of passenger or context-dependent mutations. This pattern underscores the temporal dynamics of driver acquisition in tumorigenesis [11].
The gradual accumulation of point mutations throughout a lifespan in both healthy tissues [13–17] and cancers [18] highlights the critical need to distinguish somatic from germline events to inform precision oncology [19].
CarcSeq: Design and Methodology
To address this, our group developed an error-corrected NGS method, CarcSeq, designed to detect a curated panel of early recurrent somatic point mutations in oncogenes and tumor suppressor genes [8]. CarcSeq employs multiple high-fidelity PCR reactions to amplify target loci, with each amplicon tagged by a 9-base-pair unique molecular identifier. Sequenced libraries are processed bioinformatically to generate single-strand consensus sequences (Figure 1), allowing accurate quantification of both mutant and wild-type alleles.
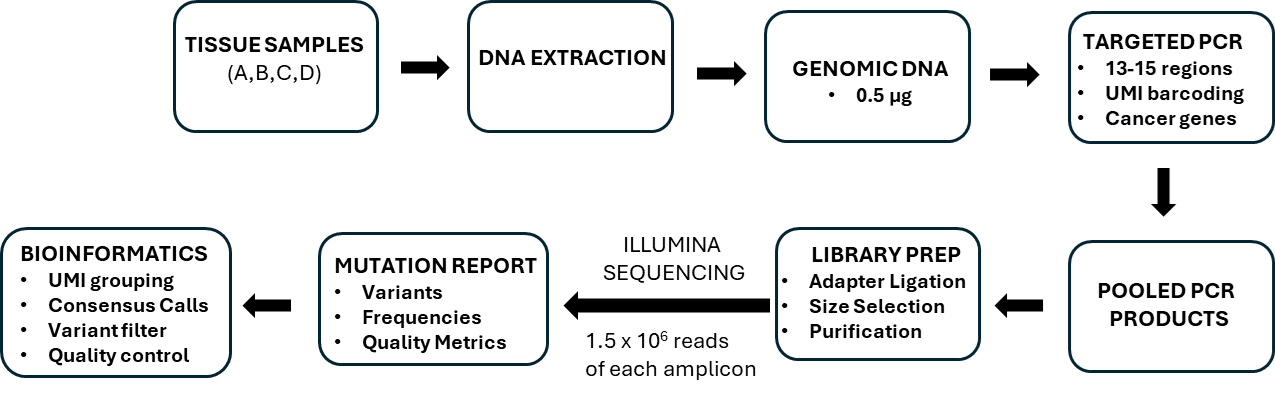
Figure 1. Workflow of methods used for CarcSeq.
While mutant fraction (MF) provides a sensitive measure of rare variant abundance, MF alone does not fully capture the heterogeneity or dispersion of mutation burdens across genomic loci or biological replicates—features that are critical for inferring early clonal expansion. To address this limitation, we incorporated the median absolute deviation (MAD) of MF as a metric. MAD provides a robust, distribution-agnostic measure of variability that is less influenced by extreme values than variance-based statistics [20], making it particularly well-suited for low-frequency mutation data generated by error-corrected NGS. As described below, MAD emerged as a sensitive indicator of clonal expansion and neoplastic potential across species and tissue types.
Application of CarcSeq in Human Tissues
In initial studies, CarcSeq was applied to 13 gene regions encompassing more than 20 hotspot CDMs in DNA from normal human breast and lung tissues, as well as matched breast and lung tumors. Raw sequencing output (mutpos files) was processed using a three-step filtering pipeline as described in Harris et al. [8]. First, mutant fractions (MFs) supported by only one or two mutant single-strand consensus sequences (SSCSs) were excluded to eliminate imprecise estimates arising from rare-event sampling error [21]. For each mutation class (base substitutions to T, C, G, or A, as well as insertions and deletions), MF was calculated as the number of mutant SSCSs (≥3) divided by the effective sequencing depth, defined as the total number of SSCSs minus those lacking a confident error-corrected base call (Ns; positions where no single base was represented by ≥90% of SSCSs). Using this approach, CarcSeq quantified mutations with a sensitivity of 10-4 and revealed tissue-specific mutational profiles, including broader mutation distributions in normal breast tissue compared with ductal carcinomas, whereas tumors exhibited higher-frequency mutations concentrated at known hotspots [8].
Application of CarcSeq in Rodent Models
Leveraging this sensitivity, we adapted CarcSeq for rodent models to assess clonal expansion and neoplastic potential by distinguishing nonsynonymous mutations (altering the encoded amino acid) from synonymous mutations (silent substitutions). In mammary tissue from 16-week-old untreated Fischer 344, Wistar Han, and Sprague Dawley rats—strains with varying susceptibility to spontaneous mammary neoplasia—CarcSeq detected hundreds of recurrent mutants (≥10-4) per strain, with 42.5% of nonsynonymous variants mapping to human homologs. Sprague Dawley rats, the most tumor-prone strain, showed elevated nonsynonymous/synonymous mutation ratios in Hras, Pik3ca, and Tp53, consistent with positive selection and clonal expansion. The MAD in these loci correlated perfectly with spontaneous mammary tumor incidence at 104 weeks (r = 1.0, p = 0.001), supporting CarcSeq-derived mutation metrics as early predictors of neoplastic outcomes [22].
We next established a mouse CarcSeq panel to quantify low-frequency mutations across hotspot CDMs (Harris et al., 2021). In lung DNA from B6C3F1 and CD-1 mice, CarcSeq recovered 1,586 recurrent mutants (≥10-4), with 55.5% of nonsynonymous variants overlapping with human tumor mutations, including hotspot codons. Male B6C3F1 mice, the most lung tumor–sensitive model, exhibited the highest nonsynonymous/synonymous ratio, consistent with positive selection. The MAD in Braf, Egfr, Kras, Stk11, and Tp53 correlated with spontaneous lung tumor incidence at 2 years of age. Similarly, human lung DNA showed borderline associations between MAD, age, and cumulative lung cancer risk. Together, these findings highlight MAD as an early metric of clonal expansion and a candidate cross-species biomarker of neoplastic susceptibility [23].
Detection of Mutations Induced by Nongenotoxic Exposures
Once CarcSeq validation was confirmed in both humans and rodents, the next step was to assess the assay’s ability to detect mutations produced by nongenotoxic exposures. Lorcaserin, a selective 5-hydroxytryptamine 2C (5-HT2C) receptor agonist and nongenotoxic carcinogen that induces mammary tumors in rats, was administered to female Sprague Dawley at doses of 0, 30, or 100 mg/kg lorcaserin daily by oral gavage for 12 or 24 weeks. Importantly, the potential genotoxic impurity N-nitroso-lorcaserin was not detected by quantitative liquid chromatography with tandem mass spectrometry (LC–MS/MS) analyses of dosing solutions, plasma, liver, or mammary tissue, supporting the classification of lorcaserin as a nongenotoxic carcinogen. Mammary DNA was interrogated for hotspot driver mutations in Apc, Braf, Egfr, Hras, Kras, Nfe2l2, Pik3ca, Setbp1, Stk11, and Tp53, with MFs determined using the CarcSeq platform [24]. No overall differences in MF were detected across the lorcaserin dose groups; however, significant dose-dependent increases in Pik3ca H1047R mutations were observed at both 12 and 24 weeks, with higher mutant counts and mutant fractions occurring at 24 weeks. These findings indicate that lorcaserin promotes clonal expansion of spontaneous Pik3ca H1047R mutants, contributing to mammary carcinogenesis. The study further establishes CarcSeq as a sensitive approach for detecting early clonal expansion events induced by nongenotoxic carcinogens within short treatment durations [24].
The studies summarized above set the foundation for our most recent published work [25]. This study utilized the sex-related differences in lung tumor susceptibility of rasH2-Tg mice to optimize analytical strategies for quantifying clonal expansion using CarcSeq. MAD revealed significantly greater clonal expansion in the male mice, particularly when calculated from lung-specific driver mutations. Males exhibited more recurrent and phenotypically relevant mutations, supporting this interpretation. These findings validated the MAD in MF as a biomarker of clonal expansion and provided methodological guidance for cancer driver gene selection and data normalization [25]. This study further validates CarcSeq quantification of MF and MAD in MF as biomarkers of clonal expansion and potential predictors of cancer risk.
Comparison of CarcSeq with Other NGS Methods
A comparison of CarcSeq with whole-genome sequencing, whole-exome sequencing, duplex sequencing, and Safe-SeqS is provided in Table 1 [8,26–29]. Conventional next-generation sequencing approaches such as whole-genome and whole-exome sequencing are optimized for comprehensive mutation discovery but lack the sensitivity required to reliably detect low-frequency somatic variants, particularly in normal tissues, due to intrinsic sequencing and PCR error rates. Although these methods are well suited for identifying clonal mutations in established tumors, variants present below approximately 1–5% VAF are often indistinguishable from technical artifacts [30,31].
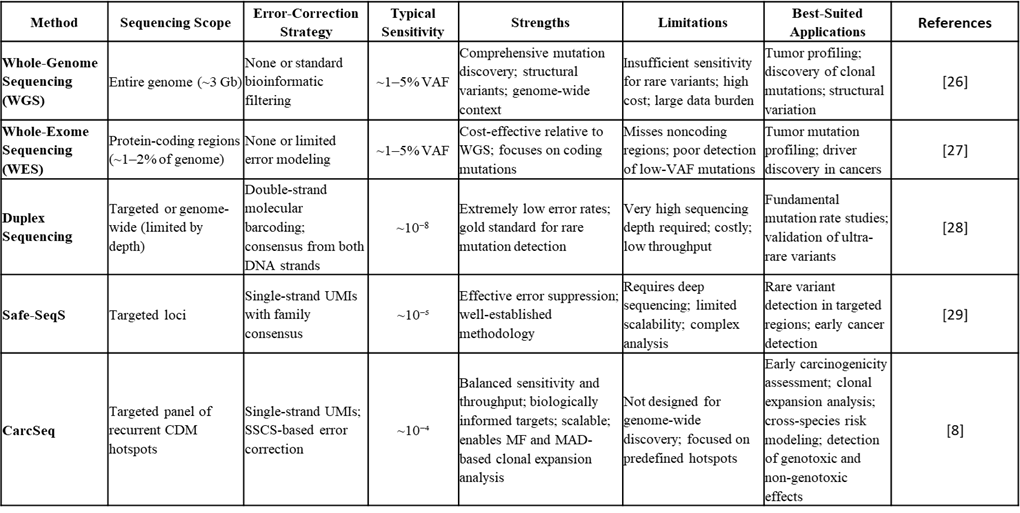
Table 1. Comparison of next-generation sequencing methods for detecting low-frequency somatic mutation.
Error-corrected sequencing methods, including duplex sequencing and Safe-SeqS, substantially improve analytical sensitivity through molecular barcoding and consensus sequencing [32,33]. Duplex Sequencing is a highly accurate DNA sequencing method that tags and sequences both strands of a double-stranded DNA molecule, creating "families" of reads from the original molecule, then comparing them to filter out sequencing errors and artifacts achieving significantly low error rates (>1x10-8) in detecting rare mutations. Duplex Sequencing has been employed for mutagenesis detection and quantification because it reports only mutations present on both strands of the same DNA molecule, ensuring that observed variants represent true, fixed mutations rather than transient DNA lesions or sequencing artifacts. [34].
Safe-SeqS provides robust reduction of PCR and sequencing errors through single-strand molecular barcoding and consensus calling, enabling sensitive detection of low-frequency variants (10-5-10-6) with relatively modest sequencing depth, high molecular recovery, and simple library preparation [34]. Safe-SeqS is also utilized for the detection of mutagenesis by identifying increases in low-frequency single-nucleotide variants and small insertions or deletions across targeted regions following exposure to a mutagen. Its single-strand consensus approach substantially reduces technical errors, allowing statistically robust comparisons of mutation frequency and spectrum between treated and control samples, which makes it suitable for higher-throughput mutagenesis studies [35,36].
CarcSeq occupies a complementary niche by combining targeted, biologically informed selection of recurrent cancer driver mutation hotspots with single-strand consensus sequencing to achieve its sensitivity (~10-4). CarcSeq is designed to quantify biologically relevant low-frequency mutations and their dispersion across loci. By enabling analysis of MF and median absolute deviation, CarcSeq provides a quantitative framework for assessing early clonal expansion and neoplastic potential, making it particularly well-suited for translational carcinogenicity assessment and cross-species risk modeling [8].
Current Developments in Sequencing Technologies for Carcinogenesis
Recent advances in sequencing technologies are transforming the detection and characterization of early carcinogenic events. Error-corrected next-generation sequencing (ecNGS) methods—including duplex sequencing, Safe-SeqS, and molecular-barcoded approaches—have dramatically reduced technical errors, enabling detection of rare somatic variants at frequencies as low as 10-5-10-6. These approaches improve resolution of low-frequency driver mutations that are critical for assessing early clonal expansion in both normal and pre-neoplastic tissues [37,38].
Single-cell sequencing and spatial transcriptomics are expanding the ability to map mutational landscapes at cellular resolution, linking clonal dynamics to tissue architecture. By capturing heterogeneity across individual cells, these methods provide insight into the temporal and spatial progression of somatic mutations, clonal selection, and microenvironmental influences on tumor initiation. In parallel, ultra-deep targeted panels enable high-throughput profiling of known cancer driver mutations across large cohorts, balancing sensitivity with cost and scalability [39,40].
Computational developments complement experimental advances. Machine learning–based variant callers and consensus-calling algorithms reduce false positives and enhance the predictive accuracy of low-frequency variant detection. Integrating longitudinal sampling with these bioinformatic tools allows the reconstruction of clonal evolution over time, facilitating the identification of early biomarkers predictive of neoplastic risk [41].
Additionally, multi-omic approaches are emerging, where mutation detection is integrated with epigenomic, transcriptomic, and proteomic profiling. This holistic view enables identification of functional consequences of low-frequency mutations, linking genomic alterations to changes in gene expression, pathway activation, and early cellular transformation [38]. Together, these technological developments promise increasingly quantitative, predictive, and mechanistically informative platforms for assessing carcinogenic potential, guiding preclinical studies, and informing translational and regulatory decision-making.
Discussion
CarcSeq provides a sensitive, scalable approach for detecting low-frequency cancer driver mutations across human and rodent tissues. By combining single-strand consensus sequencing with a targeted panel of recurrent CDM hotspots, the method achieves detection limits (~10-4) that surpass conventional whole-genome and whole-exome sequencing, which are constrained by PCR and sequencing error rates. Importantly, the application of MAD to MF data captures both the prevalence and heterogeneity of mutations, enabling robust assessment of early clonal expansion in normal and tumor-adjacent tissues.
Application of CarcSeq in rodent models demonstrated that MAD correlates with spontaneous tumor incidence and highlights strain- and tissue-specific patterns of clonal expansion. Similarly, studies of nongenotoxic exposures, such as lorcaserin, revealed dose-dependent expansion of Pik3ca H1047R mutants, confirming that CarcSeq can detect subtle early neoplastic events that traditional genotoxicity assays might miss [24]. These findings support MAD as a quantitative, cross-species biomarker of neoplastic susceptibility and reinforce CarcSeq’s utility for translational and preclinical carcinogenicity assessment.
Despite its strengths, CarcSeq is inherently targeted and not intended for de novo mutation discovery, and the predictive value of MAD for human cancer risk requires further validation in longitudinal studies. Future work should expand its application across genotoxic and nongenotoxic agents and explore integration with epidemiological and clinical datasets.
Declarations
This manuscript reflects the views of its author and does not necessarily reflect those of the U.S. Food and Drug Administration. Any mention of commercial products is for clarification only and is not intended as approval, endorsement, or recommendation.
Funding
This work was funded by the U.S. Food and Drug Administration.
References
2. Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, et al. Somatic mutant clones colonize the human esophagus with age. Science. 2018 Nov 23;362(6417):911–7.
3. Dan S, Yuan Y, Wang Y, Chen C, Gao C, Yu S, et al. Non-Invasive Prenatal Diagnosis of Lethal Skeletal Dysplasia by Targeted Capture Sequencing of Maternal Plasma. PLoS One. 2016 Jul 19;11(7):e0159355.
4. Lim JS, Kim WI, Kang HC, Kim SH, Park AH, Park EK, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med. 2015 Apr;21(4):395–400.
5. Spence JM, Spence JP, Abumoussa A, Burack WR. Ultradeep analysis of tumor heterogeneity in regions of somatic hypermutation. Genome Med. 2015 Mar 12;7(1):24.
6. Carlson CA, Kas A, Kirkwood R, Hays LE, Preston BD, Salipante SJ, et al. Decoding cell lineage from acquired mutations using arbitrary deep sequencing. Nat Methods. 2011 Nov 27;9(1):78–80.
7. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013 Mar;31(3):213–9.
8. Harris KL, Walia V, Gong B, McKim KL, Myers MB, Xu J, et al. Quantification of cancer driver mutations in human breast and lung DNA using targeted, error-corrected CarcSeq. Environ Mol Mutagen. 2020 Nov;61(9):872–89.
9. Myers MB, McKinzie PB, Wang Y, Meng F, Parsons BL. ACB-PCR quantification of somatic oncomutation. InMolecular Toxicology Protocols 2014 Feb 4 (pp. 345–363). Totowa, NJ: Humana Press.
10. Gregory MT, Bertout JA, Ericson NG, Taylor SD, Mukherjee R, Robins HS, et al. Targeted single molecule mutation detection with massively parallel sequencing. Nucleic acids research. 2016 Feb 18;44(3):e22-.
11. Gerstung M, Jolly C, Leshchiner I, Dentro SC, Gonzalez S, Rosebrock D, et al. The evolutionary history of 2,658 cancers. Nature. 2020 Feb;578(7793):122–8.
12. Campbell PJ, Getz G, Stuart JM, Korbel JO, Stein LD. ICGC/TCGA Pan-Cancer analysis of whole genomes net. Pan-cancer analysis of whole genomes. bioRxiv. 2018:1–29.
13. Moore L, Leongamornlert D, Coorens TH, Sanders MA, Ellis P, Dentro SC, et al. The mutational landscape of normal human endometrial epithelium. Nature. 2020 Apr 30;580(7805):640–6.
14. Lee-Six H, Olafsson S, Ellis P, Osborne RJ, Sanders MA, Moore L, et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature. 2019 Oct 24;574(7779):532–7.
15. Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015 May 22;348(6237):880–6.
16. Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012 Jul 20;150(2):264–78.
17. Yokoyama A, Kakiuchi N, Yoshizato T, Nannya Y, Suzuki H, Takeuchi Y, et al. Age-related remodelling of oesophageal epithelia by mutated cancer drivers. Nature. 2019 Jan 17;565(7739):312–7.
18. Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, et al. Clock-like mutational processes in human somatic cells. Nature genetics. 2015 Dec;47(12):1402–7.
19. Cheng J, Cabezon FA, Que Y, Schinckel AP. 071 Evaluation of the impact of the magnitude of errors in the sorting of pigs for market on the optimal market weight. Journal of Animal Science. 2017 Mar 1;95(suppl_2):33–4.
20. Douglas Zhang X, Yang XC, Chung N, Gates A, Stec E, Kunapuli P, et al. Robust statistical methods for hit selection in RNA interference high-throughput screening experiments. Pharmacogenomics. 2006 Apr 1;7(3):299–309.
21. McKinzie PB, Delongchamp RR, Heflich RH, Parsons BL. Prospects for applying genotypic selection of somatic oncomutation to chemical risk assessment. Mutation Research/Reviews in Mutation Research. 2001 Oct 1;489(1):47–78.
22. McKim KL, Myers MB, Harris KL, Gong B, Xu J, Parsons BL. CarcSeq measurement of rat mammary cancer driver mutations and relation to spontaneous mammary neoplasia. Toxicological Sciences. 2021 Jul 1;182(1):142–58.
23. Harris KL, McKim KL, Myers MB, Gong B, Xu J, Parsons BL. Assessment of clonal expansion using CarcSeq measurement of lung cancer driver mutations and correlation with mouse strain-and sex-related incidence of spontaneous lung neoplasia. Toxicological Sciences. 2021 Nov 1;184(1):1–4.
24. Faske JB, Myers MB, Bryant M, He X, McLellen F, Bourcier T, et al. CarcSeq detection of lorcaserin-induced clonal expansion of Pik3ca H1047R mutants in rat mammary tissue. Toxicological Sciences. 2024 Sep 1;201(1):129–44.
25. Harris KL, Faske JB, Gong B, Parsons BL. Tissue and Sex-Specific Performance of a Cancer Driver Based Biomarker in rasH2-Tg Mice. Environmental and molecular mutagenesis. 2025 Aug 13;66(8):463.
26. Park ST, Kim J. Trends in next-generation sequencing and a new era for whole genome sequencing. International neurourology journal. 2016 Nov 22;20(Suppl 2):S76.
27. Tetreault M, Bareke E, Nadaf J, Alirezaie N, Majewski J. Whole-exome sequencing as a diagnostic tool: current challenges and future opportunities. Expert review of molecular diagnostics. 2015 Jun 3;15(6):749–60.
28. Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, Loeb LA. Detection of ultra-rare mutations by next-generation sequencing. Proceedings of the National Academy of Sciences. 2012 Sep 4;109(36):14508–13.
29. Menon V, Brash DE. Next-generation sequencing methodologies to detect low-frequency mutations:“Catch me if you can”. Mutation Research-Reviews in Mutation Research. 2023 Jul 1;792:108471.
30. Clark MJ, Chen R, Lam HY, Karczewski KJ, Chen R, Euskirchen G, et al. Performance comparison of exome DNA sequencing technologies. Nature biotechnology. 2011 Oct;29(10):908–14.
31. Gundry M, Vijg J. Direct mutation analysis by high-throughput sequencing: from germline to low-abundant, somatic variants. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2012 Jan 3;729(1-2):1–5.
32. Kennedy SR, Schmitt MW, Fox EJ, Kohrn BF, Salk JJ, Ahn EH, et al. Detecting ultralow-frequency mutations by Duplex Sequencing. Nature protocols. 2014 Nov;9(11):2586–606.
33. Salk JJ, Schmitt MW, Loeb LA. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nature Reviews Genetics. 2018 May;19(5):269–85.
34. Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proceedings of the National Academy of Sciences. 2011 Jun 7;108(23):9530–5.
35. Ståhlberg A, Krzyzanowski PM, Jackson JB, Egyud M, Stein L, Godfrey TE. Simple, multiplexed, PCR-based barcoding of DNA enables sensitive mutation detection in liquid biopsies using sequencing. Nucleic acids research. 2016 Jun 20;44(11):e105.
36. Salk JJ, Schmitt MW, Loeb LA. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nature Reviews Genetics. 2018 May;19(5):269-85.
37. Koh G, Zou X, Nik-Zainal S. Mutational signatures: experimental design and analytical framework. Genome biology. 2020 Feb 14;21(1):37.
38. Saviano A, Henderson NC, Baumert TF. Single-cell genomics and spatial transcriptomics: discovery of novel cell states and cellular interactions in liver physiology and disease biology. Journal of hepatology. 2020 Nov 1;73(5):1219–30.
39. Zhang X, Chu C, Zhang Y, Wu Y, Gao J. Concod: Accurate consensus-based approach of calling deletions from high-throughput sequencing data. In2016 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) 2016 Dec 15 (pp. 72-77). IEEE.
40. Lim J, Park C, Kim M, Kim H, Kim J, Lee DS. Advances in single-cell omics and multiomics for high-resolution molecular profiling. Experimental & molecular medicine. 2024 Mar;56(3):515–26.
41. Kukita Y, Matoba R, Uchida J, Hamakawa T, Doki Y, Imamura F, et al. High-fidelity target sequencing of individual molecules identified using barcode sequences: de novo detection and absolute quantitation of mutations in plasma cell-free DNA from cancer patients. DNA Research. 2015 Aug 1;22(4):269–77.