Commentary
FBXO9, an evolutionarily conserved member of the F-box protein family, functions as a substrate receptor within the Cullin 1-RING ubiquitin ligase complex (CRL1), also known as the SKP1-Cullin 1-F-box (SCF) complex [1]. The CRL1/SCF complex is crucial for the ubiquitin-proteasome system, which governs protein degradation and maintains cellular homeostasis. Dysregulation of this system has been implicated in cancer pathogenesis [2]. Much of the prior research has focused on well-known F-box proteins, such as SKP2, β-TrCP, and FBXW7. These proteins play essential roles in regulating cell cycle progression, apoptosis, and DNA damage response and have been linked to tumorigenesis. For instance, SKP2 is vital for the degradation of p27, a cyclin-dependent kinase inhibitor that facilitates cell cycle progression. Similarly, β-TrCP targets substrates such as IκBα, an inhibitor of the NF-κB signaling pathway, whereas FBXW7 regulates critical oncogenes, such as MYC and cyclin E2 [1-3]. In contrast, FBXO9 has been less extensively studied, despite its involvement in the CRL1/SCF complex. This represents a largely untapped opportunity for therapeutic intervention in cancer treatment. Recent studies have begun to uncover the multifaceted roles of FBXO9, addressing significant gaps in our understanding of its importance in cancer biology.
One notable aspect of the F-box protein FBXO9 is its variable expression pattern across different types of cancer. Elevated levels of FBXO9 are associated with oncogenic activities in multiple myeloma [4] and hepatocellular carcinoma [5], whereas reduced levels are correlated with the pathogenesis of acute leukemia [6]. Recent findings [7] from our laboratory indicate that FBXO9 levels are significantly lower in lung cancer tissues compared to adjacent normal tissues. Statistical analyses using Kaplan-Meier plots demonstrate a direct correlation between low FBXO9 levels and poor patient outcomes. Notably, the restoration of FBXO9 expression in lung cancer cells significantly inhibits metastatic behaviors, including cell migration, tumor-initiating cell spheroid formation, and overall metastasis. Our findings align with extant research recognizing that F-box proteins can function as either oncogenes or tumor suppressors depending on the context. For instance, β-TrCP operates as a tumor suppressor in cancers such as colorectal cancer and hepatoblastoma while promoting tumor growth in gastric, prostate, and mammary cancers [8]. Our data, corroborated by other studies, underscore the underexpression of FBXO9 in lung cancer in contrast to its overexpression in cancers such as multiple myeloma [4] and hepatocellular carcinoma [5]. This emphasizes the importance of cellular context and the microenvironment in determining the role of FBXO9. These findings suggest a complex regulatory mechanism that warrants further elucidation through carefully controlled, context-specific functional assays. Considering FBXO9’s pivotal role in the CRL1/SCF complex, understanding its differential expression across various cancers could offer novel therapeutic insights. Comprehensive analyses of tumors from The Cancer Genome Atlas (TCGA) using TIMER software (https://cistrome.shinyapps.io/timer/) revealed its underexpression in several other solid tumors besides lung cancer, including breast cancer, colorectal cancer, and chromophobe renal cell carcinoma, juxtaposed with its upregulation in liver hepatocellular carcinoma and cholangiocarcinoma (Figure 1). This dichotomy raises significant questions regarding FBXO9’s role as either a tumor promoter or inhibitor, likely varying across tissue types, disease stages, and cellular contexts. Understanding these differences is crucial for developing effective targeted therapies.
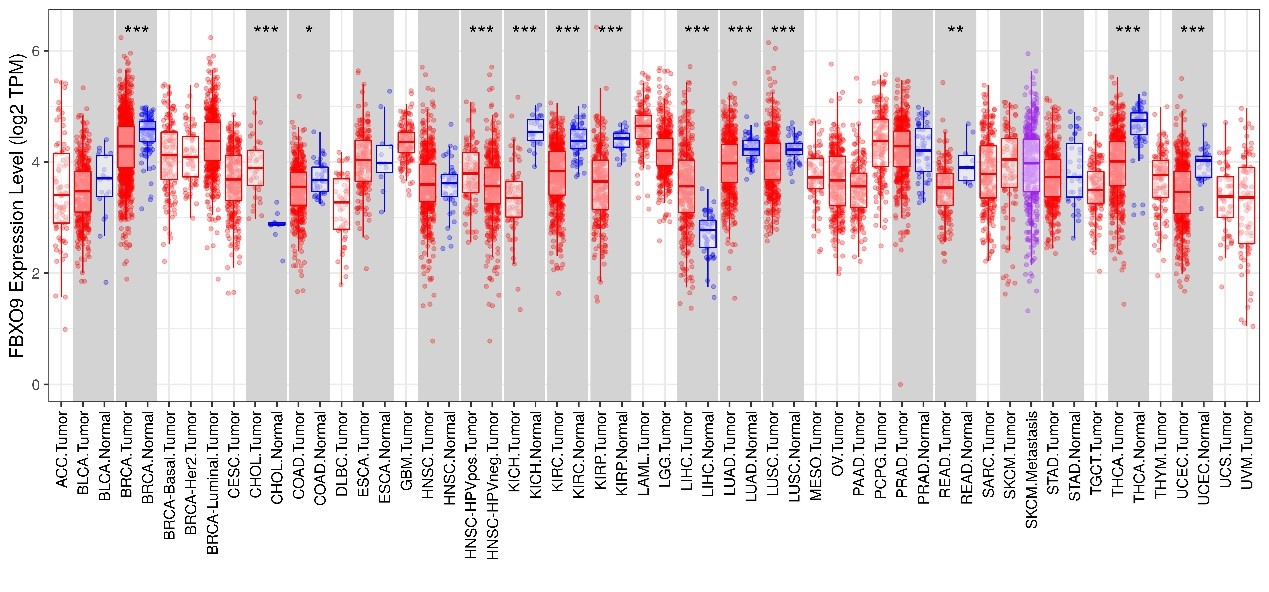
Figure 1: Differential expression of FBXO9 in normal and tumor tissues across various cancers based on data from the TIMER website.
The clinical importance of lung cancer underscores the need to scrutinize the role of FBXO9, particularly because of its markedly reduced expression and association with a poor prognosis. Our research [7] has demonstrated that FBXO9 exhibits anticancer properties in lung cancer, primarily through its involvement in the CRL1/SCF ligase complex. In contrast to earlier claims that FBXO9 exerts its effects via proteasome inhibition in acute leukemia [6], our study [7] uncovered a novel mechanism for FBXO9. Specifically, FBXO9 inhibits vacuolar-type H+-ATPase (V-ATPase), a crucial regulator of the intracellular pH integral to cancer pathology [9], by inducing non-proteolytic ubiquitination of the ATP6V1A catalytic subunit within the V1 domain of V-ATPase. This post-translational modification enhances the binding of ATP6V1A to the cytoplasmic chaperone HSPA8, sequestering ATP6V1A in the cytoplasm, and preventing the assembly of functional V-ATPase complexes on the cellular membrane. This discovery significantly enhances our understanding of the regulation of V-ATPase activity through FBXO9-mediated ubiquitination. It complements the effects of PKA- and AMPK-mediated phosphorylation and glycosylation of the V-ATPase V1 and Vo domains, which promote the enzyme’s activity within cells [10-12]. FBXO9 disruption of the vesicular acidification is critical for metastatic progression, thereby inhibiting metastasis-related cellular behaviors, such as in vitro migration and neoplastic spheroid formation, as well as reducing overall metastasis in a mouse model. In contrast, the absence of FBXO9 leads to increased V-ATPase assembly, vesicular acidification, and activation of oncogenic pathways, such as Wnt signaling and epithelial-mesenchymal transition (EMT), driving metastasis. This unique regulatory mechanism of V-ATPase, mediated through a ubiquitin ligase substrate receptor, highlights the sophisticated capacity of cellular systems to regulate physiological processes that contribute to disease progression when disrupted. Applying this cellular paradigm to an in vivo lung cancer mouse model, along with concomitant analyses correlating FBXO9 expression with patient prognosis, extends the boundaries of experimental oncology and demonstrates its significant clinical relevance. These findings not only validate our laboratory results but also underscore the potential therapeutic benefits of modulating V-ATPase through FBXO9 or by modifying its associated pathways. The innovative strategy of augmenting FBXO9 levels presents a novel approach for addressing the multifaceted challenges of lung cancer management. Despite the inherent complexities and substantial validation needed for the transition from experimental research to clinical applications, this study lays a promising foundation for developing treatments aimed at mitigating the invasive nature of lung cancer. A notable limitation of our study is its reliance on the tail vein injection tumor metastasis model, which does not fully capture the natural progression of metastasis. This model, while providing useful initial insights, often bypasses critical steps in the metastatic cascade, such as intravasation and extravasation, which are essential for a thorough understanding of metastatic biology. Consequently, future research should focus on elucidating the function of FBXO9 in vivo using genetically engineered mouse models. These models can be tailored to recapitulate the human disease more accurately, offering a more comprehensive understanding of the metastatic process and the role of FBXO9 within it. Moreover, research should aim to unravel the specific molecular mechanisms underpinning FBXO9’s context-dependent roles through comprehensive in vitro and in vivo studies. This involves investigating how FBXO9 interacts with other signaling molecules and pathways, and whether these interactions vary across different cancer types and stages of disease progression. The development of targeted FBXO9 agonists or gene therapy strategies to enhance FBXO9 expression could lead to effective lung cancer treatments. Small molecule screens to identify FBXO9 activators or stabilizers could pave the way for drug development. Furthermore, advanced gene therapy techniques, such as adeno-associated virus (AAV) vectors, could be utilized for targeted delivery of FBXO9. Nevertheless, it is essential to ensure the safety and efficacy of these strategies through rigorous prospective clinical trials. Such advancements have the potential to revolutionize therapeutic interventions for lung cancer patients, ultimately improving their prognosis and survival rates.
In conclusion, our study elucidates a novel function of FBXO9 in lung cancer, expanding our understanding of its anticancer mechanisms through the modulation of V-ATPase activity, beyond its recognized role in proteasome inhibition. Future research should focus on robust in vivo models, the clarification of specific molecular mechanisms, and the development of effective therapeutic strategies. This deeper insight into FBXO9’s role across different cancer types and biological contexts augments our understanding of how disruptions in these regulatory processes contribute to disease progression. This knowledge holds the potential to identify new therapeutic targets, ultimately improving patient outcomes.
Funding
This work is partially supported by the National Natural Science Foundation of China (Grant 81872393 and 81472793).
Declaration of Competing Interest
There is no conflict of interest.
References
2. Yumimoto K, Yamauchi Y, Nakayama KI. F-Box Proteins and Cancer. Cancers (Basel). 2020 May 15;12(5):1249.
3. Wang Z, Liu P, Inuzuka H, Wei W. Roles of F-box proteins in cancer. Nat Rev Cancer. 2014 Apr;14(4):233-47.
4. Fernández-Sáiz V, Targosz BS, Lemeer S, Eichner R, Langer C, Bullinger L, et al. SCFFbxo9 and CK2 direct the cellular response to growth factor withdrawal via Tel2/Tti1 degradation and promote survival in multiple myeloma. Nat Cell Biol. 2013 Jan;15(1):72-81.
5. Wang Z, Chen X, Zhou L, Zhao X, Ge C, Zhao F, et al. FBXO9 Mediates the Cancer-Promoting Effects of ZNF143 by Degrading FBXW7 and Facilitates Drug Resistance in Hepatocellular Carcinoma. Front Oncol. 2022 Jun 30;12:930220.
6. Hynes-Smith RW, Swenson SA, Vahle H, Wittorf KJ, Caplan M, Amador C, et al. Loss of FBXO9 Enhances Proteasome Activity and Promotes Aggressiveness in Acute Myeloid Leukemia. Cancers (Basel). 2019 Nov 3;11(11):1717.
7. Liu L, Chen X, Wu L, Huang K, Wang Z, Zheng Y, et al. Ubiquitin ligase subunit FBXO9 inhibits V-ATPase assembly and impedes lung cancer metastasis. Exp Hematol Oncol. 2024 Mar 14;13(1):32.
8. Bi Y, Cui D, Xiong X, Zhao Y. The characteristics and roles of β-TrCP1/2 in carcinogenesis. FEBS J. 2021 Jun;288(11):3351-74.
9. Stransky L, Cotter K, Forgac M. The Function of V-ATPases in Cancer. Physiol Rev. 2016 Jul;96(3):1071-91.
10. Cotter K, Stransky L, McGuire C, Forgac M. Recent Insights into the Structure, Regulation, and Function of the V-ATPases. Trends Biochem Sci. 2015 Oct;40(10):611-22.
11. Eaton AF, Merkulova M, Brown D. The H+-ATPase (V-ATPase): from proton pump to signaling complex in health and disease. Am J Physiol Cell Physiol. 2021 Mar 1;320(3):C392-C414.
12. Lee MR, Lee GH, Lee HY, Kim DS, Chung MJ, Lee YC, et al. BAX inhibitor-1-associated V-ATPase glycosylation enhances collagen degradation in pulmonary fibrosis. Cell Death Dis. 2014 Mar 13;5(3):e1113.