Abstract
Prostate cancer (Pca) is one of the most common cancers among men worldwide. The current screening methods lack effectiveness such as prostate-specific antigen (PSA) and Magnetic resonance imaging (MRI), and some others come with pain such as biopsy. Understanding the genomic behavior of the disease may play a key part in designing more effective, accurate, and less invasive diagnosis measures. Pca has many clinical features to describe the spread and the aggressiveness of the tumor including Gleason score, TNM staging system, and the location of the tumor in the prostate gland which is known as laterality.
Machine learning models were recently utilized to predict the outcomes of Pca, and to find potential biomarkers for the clinical features of the disease. In this study, we review recent machine learning methods for finding biomarkers for Pca clinical features including Pca progression, Gleason score, and laterality. The supervised models were built on gene expressions and next-generation sequencing data to find genes or genes transcripts that are associated with these clinical features. The results show high performance in the three models with an accuracy of more than 90%. The three models reported many biomarkers genes and genes transcripts including but not restricted to CARNA22, DOCK9, FLVCR2, IK2F3, USP13, PTGFR, and CLASP1 genes for Pca progression. UBE2V2, GPR137, and EPB41L1 for different Gleason scores. And FBXO21, RTN1, NDUFA5, ALG5, Z99129, SNAI2, MRI1, HLA-DMB, SRSF6, and EIF4G2 for laterality prediction.
Keywords: Machine learning, prostate cancer diagnosis, next-generation sequencing, gene expression.
Keywords
Machine learning, prostate cancer diagnosis, next-generation sequencing, gene expression.
Introduction
Pca screening models are not efficient in the diagnosis of the disease. Alanee et al. reported 33 patients out of 156 who underwent prostatectomy for Pca Gleason ≥ 3+4 diagnosed on prostate biopsy had negative MRI results [1]. PSA level which can be measured in a blood test with less pain than biopsy is found to save much life by early detecting cancer. However, not every high level of PSA means the existence of Pca, and some Pca patients may not show a high level of PSA in their blood test [2,3]. TNM staging system describes the amount and spread of cancer in a patient’s body. T describes the size of the tumor and any spread of cancer into nearby tissue; N describes the spread of cancer to nearby lymph nodes; M describes metastasis (spread of cancer to other parts of the body) [4].
Identifying genomic biomarkers for prostate cancer (Pca) is gaining research interests due to the advances of the emerging next-generation sequencing (NGS) technology [5]. NGS provides a deep insight into the gene transcription events in cancer cells and increases the sensitivity of detecting genes relationships [6]. NGS generates a huge amount of data with some artifacts, and pre-processing the data is highly recommended [7]. Genomic data such as gene expression and RNA-Seq data provide an insight into the genomic activity in the tumor tissue which leads to a better understanding of the development of the disease.
In this communication, we are surveying three classification models to predict the outcome of the Pca using RNA-Seq data. The first model is to extract potential biomarkers for Pca progression, where the classes of the models represent different TNM stages/sub-stages [8]. The second model to predict different Gleason scores [9,10]. The third model to predict the laterality of the tumor in the prostate gland [11]. Identifying the laterality of Pca can help to determine candidates for hemiablation of the prostate using focal therapy while preserving the contralateral lobe [12]. Most of the early detected cases have the tumor in a specific location in the gland, and for these cases, focal therapy can minimize negative side effects that may result from prostatectomy and radiation. The side effects of these extreme procedures include urinary incontinence, erectile dysfunction and bowel toxicity [13]. These three models can be investigated to find biomarkers that have better accuracy, more efficient, and less invasive than the current diagnosis approaches [14].
Materials & Methods
In this communication, the methods were applied to the following datasets:
- Long et al. data set that contains RNA-Seq data for 104 samples from 100 patients. The clinical data includes information about the TNM stage and Gleason score [15].
- Kannan et al. data set RNA-Seq for 20 human prostate cancer tissues and 10 matched benign from patients who had received no preoperative therapy prior to radical prostatectomy [16].
- Prostate adenocarcinoma (TCGA-PRAD) data set that contains gene expressions for 498 prostate cancer patient samples with different Gleason scores [17].
Preprocessing
RNA-Seq datasets were preprocessed by Zseq RNA-Seq reads filter [7] first, then aligned to the human genome using Tophat2 [18] for Long and Kannan datasets to study the progression and Long datasets using STAR [19] to analyze different Gleason score biomarkers. Then Cufflinks [20] and RSEM [21] were used to construct the genes transcripts for Tophat2 and STAR outputs. Finally, the transcript constructor quantifies the reads on the transcripts to measure the expressions using transcripts per million of reads (TPM) measure. Figure 1 shows the preprocessing pipeline.

Figure 1: RNA-Seq data preprocessing pipeline.
Feature Selection
To remove any irrelevant genes transcript quantifications such as inactive genes and housekeeping genes, we applied the Information Gain (IG) feature selection method to rank the features based on the coloration with the class, then the minimum Redundancy Maximum Relevance (mRMR) wrapper method [22] was applied on the ranked genes to identify the potential biomarkers genes for the better prediction performance. Table 1 depicts the used methods on each data set for specific prostate cancer problem. mRMR is wrapped on a standard machine learning machine to achieve high-performance measurements. The utilized classifiers for each Pca problem are demonstrated in the next subsection.
|
Dataset |
Feature ranking method |
The problem |
|
Long et al dataset |
mRMR |
Pca Progression |
|
Long et al dataset |
IG and mRMR |
Pca Gleason score |
|
TCGA-PRAD |
IG and mRMR |
Pca Gleason score |
|
TCGA-PRAD |
IG and mRMR |
Pca Laterality |
Prediction Models
The preprocessed datasets are fed to the prediction models. Different machine learning classification models were utilized to find potential biomarkers genes that can predict different outcomes of Pca as the following:
Pca Progression
Multiple prediction models for each consecutive Pca stages/sub-stages were built to extract biomarkers that can predict progression in each step. Each pair of consecutive stages’ samples, namely, T2a-T2b, T2b-T2c, T2c-T3a, and T2c-T3/T4 was fed to the mRMR wrapper based on support vector machine (SVM) [23] classifier to select the transcripts that increased the accuracy of the prediction model. These discriminative transcripts can distinguish each stage/sub-stage from the earlier one.
The initial study has analyzed the transitions for the pairs of T1c-T2, T2-T2a, and T3a-T3b, but we decided to not include them in this study. According to world health organization (WHO) definition, T2 encompasses T2a, T2b, and T2c, therefore T2 to T2a does not mean disease progression. Likewise, T3a and T3b are defined as extra-prostatic invasion and seminal vesicle invasion, respectively, and T3a to T3b does not mean disease progression [24]. The transition from T1c sub-stage, which is diagnosed by needle biopsy, to T2 which is diagnosed by prostatectomy may not mean progression, because their difference is the way of sampling.
Pca Gleason Score
Unlike the pairwise Pca progression model, this model was constructed as a class versus the rest-based prediction model, where a specific Gleason score’s samples will be classified versus the rest of the samples. The greedy hierarchical model starts with the class that produces the best accuracy when it is classified versus the rest. Then the samples of the already classified class will be removed and the next node will consider the best performance class among the remaining. The process continues until it reaches 2 remaining classes down the hierarchy as seen in Figure 2. The considered Gleason scores were group based on Epstein model [25] where any score <=6 is considered as one group and named as group 6. The considered groups are 6, 3+4=7, 4+3=7, 8, and (9 and 10) are combined due to the low number of samples in each of them. Naïve Bayes [26] classifier was used with mRMR wrapper feature selection.
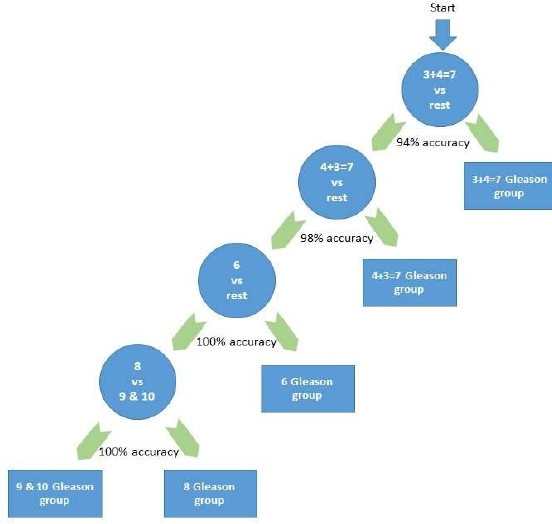
Figure 2: The hierarchical prediction model for Gleason scores (one-versus-rest) based on the quantification of the genes transcripts.
Pca Laterality
The purpose of this model is to identify the active genes that can predict the location of the tumor in the prostate gland. The model considers the three laterals which are left, right, and bi-lateral as the classes of the model. The model is constructed as one versus the others, so basically, it has 3 classifiers for each class’s samples versus the others. SVM with radial based function (RBF) kernel was used with the mRMR wrapper method.
Results
The proposed method for the Pca progression was compared to the well-known CuffDiff statistical approach which is part of Cufflinks package. The proposed model outperformed CuffDiff in all the pairwise prediction stages as seen in Table 2. In Table 2 the performance measurements are the accuracy (ACC), F-measure (FM), Matthews correlation coefficient (MCC), and area under the curve (AUC).
|
Stage |
Method |
# Selected |
# Common |
ACC |
FM |
MCC |
AUC |
|
T2A-T2B (23 vs. 11) |
CuffDiff |
35 |
0 |
64.7% |
0.601 |
0.068 |
0.634 |
|
Proposed Method |
6 |
85.3% |
0.851 |
0.657 |
0.826 |
||
|
T2B-T2C (11 vs. 30) |
CuffDiff |
38 |
0 |
65.8% |
0.647 |
0.078 |
0.645 |
|
Proposed Method |
5 |
87.8% |
0.880 |
0.699 |
0.885 |
||
|
T2C-T3A (30 vs. 8) |
CuffDiff |
29 |
0 |
73.7% |
0.722 |
0.130 |
0.612 |
|
Proposed Method |
5 |
89.4% |
0.895 |
0.683 |
0.948 |
||
|
T2C-T3/T4 (30 vs. 17) |
CuffDiff |
49 |
0 |
57.4% |
0.568 |
0.055 |
0.483 |
|
Proposed Method |
12 |
95.7% |
0.957 |
0.908 |
0.988 |
Gleason score prediction model that incorporates Naïve Bayes classifier was compared to the other standard classifier and found to outperform them. Table 3 shows the performance measurements of the proposed method.
|
Gleason Group |
ACC |
FM |
MCC |
AUC |
|
3 + 4 = 7 vs. Rest |
94 |
0.94 |
0.88 |
95 |
|
4 + 3 = 7 vs. Rest |
98 |
0.98 |
0.96 |
99 |
|
6 vs. Rest |
100 |
1 |
1 |
100 |
|
8 vs. (9 and 10) |
100 |
1 |
1 |
100 |
In the Laterality model, the results of the three classification systems to discover lateral gene biomarkers are shown in Table 4. SVM RBF outperformed Naïve Bayes and Random forest classifiers [27] in the three systems.
|
Classifier |
Accuracy |
Precision |
Accuracy |
Precision |
Accuracy |
Precision |
|
SVM RBF |
99 |
97 |
99 |
97 |
99 |
97 |
|
Naive Bayes |
88 |
78 |
82 |
78 |
80 |
78 |
|
Random Forest |
93 |
85 |
90 |
85 |
95 |
85 |
|
Prediction Model |
Left vs. rest |
Bilateral vs. rest |
Right vs. rest |
|||
The three prediction models reported many potential gene transcripts biomarkers for Pca. Pca progression model reported that the upregulation of the gene expression of the small Cajal body-specific RNA (SCARNA22) from stages T2c to T3/T4. It also reported transcripts from the genes DOCK9, FLVCR2, IK2F3, USP13, PTGFR, and CLASP1 can potentially identify the same progression.
In the second model, many gene transcripts have differentially expressed among different Gleason scores. The model revealed that transcripts of genes GPR137 and EPB41L1 is associated with tumors of Gleason scores 3+4=7 and 8, respectively. It also reported differential gene transcripts quantifications of PIAS3 and Rest Corepressor 3 (Rcor3) were both associated with tumors of Gleason score 4+3=7. A different quantification of the UBE2V2 transcript was able to predict non-advanced Pca with Gleason score 6.
The laterality model revealed some genomic activity that is related with the location of the cancer in prostate gland. Differential gene expression of FBXO21, RTN1, NDUFA5, and POP7 genes can predict the tumor in the left side of the gland. While that of HLA-DMB, SRSF6, EIF4G2 can predict it in the right side. Finally, ALG5, Z99129, SNAI2, MRI1 genes can predict the bilateral tumor.
Discussion
Finding the signature of Pca outcomes can assist in understanding the development of the disease. The identified biomarkers should be validated using wet-lab experiments, and analyze the produced proteins to design less invasive approaches such as blood or urine test. To the top of our knowledge, the three models are novel when it comes to finding signature gene biomarkers for the Pca progression, Gleason scores, and laterality. The models will bring more attention to the area of understanding of the Pca molecular based rather than the visual attributes of the tissue.
As for now, the current models are designed to predict the outcomes based on genomic profiling including gene expressions and RNA-Seq. However, the set of biomarkers can be analyzed using pathways databases and wet-lab experiments to measure the expressions or quantifications of the biomarkers in the blood or urine, which can determine the Pca outcome based on these measures. Drugs can be designed or repurposed for prescriptions to target the identified biomarkers from the three models.
Conclusion
Thanks to the advancement of NGS technology, machine learning models based on RNA-Seq data can be used to find a newer gene biomarker for Pca. These biomarkers may substitute the current Pca diagnostic methods. The genomic activity may reveal the mechanism of the Pca progression, aggressiveness, and location. The findings of the three models needs to undergo an extensive validation process using computational methods such as pathway analysis, and wet lab experiments.
Conflicts of Interest
there are no Conflicts of Interest.
Funding
This work was supported by Mitacs through the Mitacs Accelerate Program, the Natural Sciences and Engineering Research Council of Canada, NSERC, and the University of Windsor Office for Research Services and Innovation.
References
2. Mayo Clinica Staff. Prostate cancer screening: Should you get a PSA test? https://www.mayoclinic.org/tests-procedures/psa-test/in-depth/prostate-cancer/art-20048087, [Last Accessed Sep 25, 2020].
3. Saxby H, Mikropoulos C, Boussios S. An Update on the Prognostic and Predictive Serum Biomarkers in Metastatic Prostate Cancer. Diagnostics. 2020 Aug;10(8):549.
4. National Cancer Institute (NCI). https://www.cancer.gov/about-cancer/diagnosis-staging/staging. [Last Accessed Sep 25, 2020].
5. Rubin MA, Demichelis F. The Genomics of Prostate Cancer: emerging understanding with technologic advances. Modern Pathology. 2018 Jan;31(1):1-1.
6. Behjati S, Tarpey PS. What is next generation sequencing? Archives of Disease in Childhood-Education and Practice. 2013 Dec 1;98(6):236-8.
7. Alkhateeb A, Rueda L. Zseq: an approach for preprocessing next-generation sequencing data. Journal of Computational Biology. 2017 Aug 1;24(8):746-55.
8. Alkhateeb A, Rezaeian I, Singireddy S, Cavallo-Medved D, Porter LA, Rueda L. Transcriptomics signature from next-generation sequencing data reveals new transcriptomic biomarkers related to prostate cancer. Cancer informatics. 2019 Mar; 18:1176935119835522.
9. Hamzeh O, Alkhateeb A, Rezaeian I, Karkar A, Rueda L. Finding transcripts associated with prostate cancer gleason stages using next generation sequencing and machine learning techniques. In: International Conference on Bioinformatics and Biomedical Engineering 2017 Apr 26 (pp. 337-348). Springer, Cham.
10. Hamzeh O, Alkhateeb A, Zheng JZ, Kandalam S, Leung C, Atikukke G, et al. A Hierarchical Machine Learning Model to Discover Gleason Grade-Specific Biomarkers in Prostate Cancer. Diagnostics. 2019 Dec;9(4):219.
11. Hamzeh O, Alkhateeb A, Zheng J, Kandalam S, Rueda L. Prediction of tumor location in prostate cancer tissue using a machine learning system on gene expression data. BMC Bioinformatics. 2020 Mar 11; 21(Suppl 2):78.
12. Mouraviev V, Mayes JM, Sun L, Madden JF, Moul JW, Polascik TJ. Prostate cancer laterality as a rationale of focal ablative therapy for the treatment of clinically localized prostate cancer. Cancer: Interdisciplinary International Journal of the American Cancer Society. 2007 Aug 15;110(4):906-10.
13. Ahmed HU, Pendse D, Illing R, Allen C, van der Meulen JH, Emberton M. Will focal therapy become a standard of care for men with localized prostate cancer? Nature Clinical Practice Oncology. 2007 Nov;4(11):632-42.
14. Hamzeh O, Alkhateeb A, Zheng J, Kandalam S, Rueda L. Prediction of tumor location in prostate cancer tissue using a machine learning system on gene expression data. BMC Bioinformatics. 2020 Mar;21(2):1-0.
15. Long Q, Xu J, Osunkoya AO, Sannigrahi S, Johnson BA, Zhou W, et al. Global transcriptome analysis of formalin-fixed prostate cancer specimens identifies biomarkers of disease recurrence. Cancer Research. 2014 Jun 15;74(12):3228-37.
16. Kannan K, Wang L, Wang J, Ittmann MM, Li W, Yen L. Recurrent chimeric RNAs enriched in human prostate cancer identified by deep sequencing. Proceedings of the National Academy of Sciences. 2011 May 31;108(22):9172-7.
17. Prostate Adenocarcinoma TCGA-PRAD Dataset. 2020. Available online: https://portal.gdc.cancer.gov/projects/TCGA-PRAD (accessed on 29 November 2019).
18. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology. 2013 Apr 1;14(4):R36.
19. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013 Jan 1;29(1):15-21.
20. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, Van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology. 2010 May;28(5):511-5.
21. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011 Dec 1;12(1):323.
22. Peng H, Long F, Ding C. Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy. IEEE Transactions on pattern analysis and Mchine Intelligence. 2005 Jun 20;27(8):1226-38.
23. Cortes C, Vapnik V. Support-vector networks. Machine Learning. 1995 Sep 1;20(3):273-97.
24. Parker C, Gillessen S, Heidenreich A, Horwich A. Cancer of the prostate: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology. 2015 Sep 1;26(suppl 5):v69-77.
25. Gordetsky J, Epstein J. Grading of prostatic adenocarcinoma: current state and prognostic implications. Diagnostic Pathology. 2016 Dec 1; 11(1):25.
26. Domingos P, Pazzani M. On the optimality of the simple Bayesian classifier under zero-one loss. Machine Learning. 1997 Nov 1; 29(2-3):103-30.
27. Breiman L. Random forests. Machine Learning. 2001 Oct 1; 45(1):5-32.