Introduction: From Energy Production to Information Processing
The evolutionary origin of mitochondria remains a challenging problem, closely linked to the emergence of eukaryotic cells, a process known as eukaryogenesis. Regardless of how primordial mitochondria truly evolved, mitochondria have traditionally been viewed as bioenergetic organelles with primary functions of ATP production and metabolic support. They are classically known as the ‘powerhouse’ of the cells. In aging biology, this view has often been paired with an emphasis on mitochondrial dysfunction and oxidative imbalance and stress. Increasing evidence, however, supports a broader and more integrative role for mitochondria as signaling organelles responsible for communicating the cellular state to the nucleus and shape gene expression programs through metabolic and redox-dependent mechanisms [1,2].
At the center of this communication lies mitochondrial redox signaling, which operates through redox couples like NAD+/NADH, metabolic intermediates, and reactive oxygen and nitrogen species (ROS/RNS). Emerging evidence indicates that when precisely regulated, these signals function as information carriers, coordinating adaptive transcriptional responses rather than serving merely as markers of oxidative stress. This distinction becomes particularly relevant when considering the epigenome, which integrates metabolic inputs, stores regulatory memory, and governs stress-responsive gene expression [3,4].
Mitochondria are increasingly understood as doing more than producing energy. Studies have become more and more focused on they function as redox-epigenetic integrators, acting as a cellular control center that senses metabolic status and environmental conditions, such as nutrient availability and stress, and translates these signals into adaptive changes in chromatin structure and gene regulation. From this perspective, healthy aging is not defined by maintaining fixed or static molecular states, but by preserving redox-dependent epigenetic adaptability: the ability of cells to flexibly adjust gene activity over time in response to changing conditions.
Mitochondrial Outputs that Shape the Epigenome
Epigenetic regulation relies on enzymes whose activity is closely coupled to cellular metabolism. Mitochondria are uniquely positioned to influence these processes because they regulate both the availability of key metabolites and the redox environment required for chromatin remodeling (Table 1).
Mitochondrial metabolism directly controls the abundance of epigenetically active metabolites such as acetyl-CoA, α-ketoglutarate, succinate, and fumarate, which serve as substrates or regulators of histone acetylation and DNA/histone methylation and demethylation reactions. Perturbations in tricarboxylic acid (TCA) cycle flux can therefore alter chromatin states by shifting the balance between epigenetic “writing” and “erasing” activities [1,4].
Beyond metabolite supply, redox signaling modulates epigenetic machinery more directly. ROS, particularly hydrogen peroxide, can reversibly modify cysteine residues in transcription factors and chromatin-associated proteins, influencing DNA binding, cofactor recruitment, and chromatin accessibility when signaling remains spatially and temporally constrained. When these signals become chronic or diffuse, specificity is lost, and redox activity increasingly manifests as oxidative stress rather than regulatory input [1,5].
Mitochondria also regulate NAD+ homeostasis, linking redox balance to the activity of NAD+-dependent enzymes involved in chromatin remodeling, transcriptional control, and DNA repair. Age-associated declines in NAD+ availability may therefore reflect beyond metabolism, compromising the epigenetic machinery required for adaptive gene expression [6,7].
Mitochondrial redox outputs act as signaling intermediates that interface with epigenetic regulation. Precise, reversible signaling supports adaptive gene expression and resilience, whereas impaired signal fidelity promotes epigenetic dysfunction and loss of adaptive capacity (Table 1).
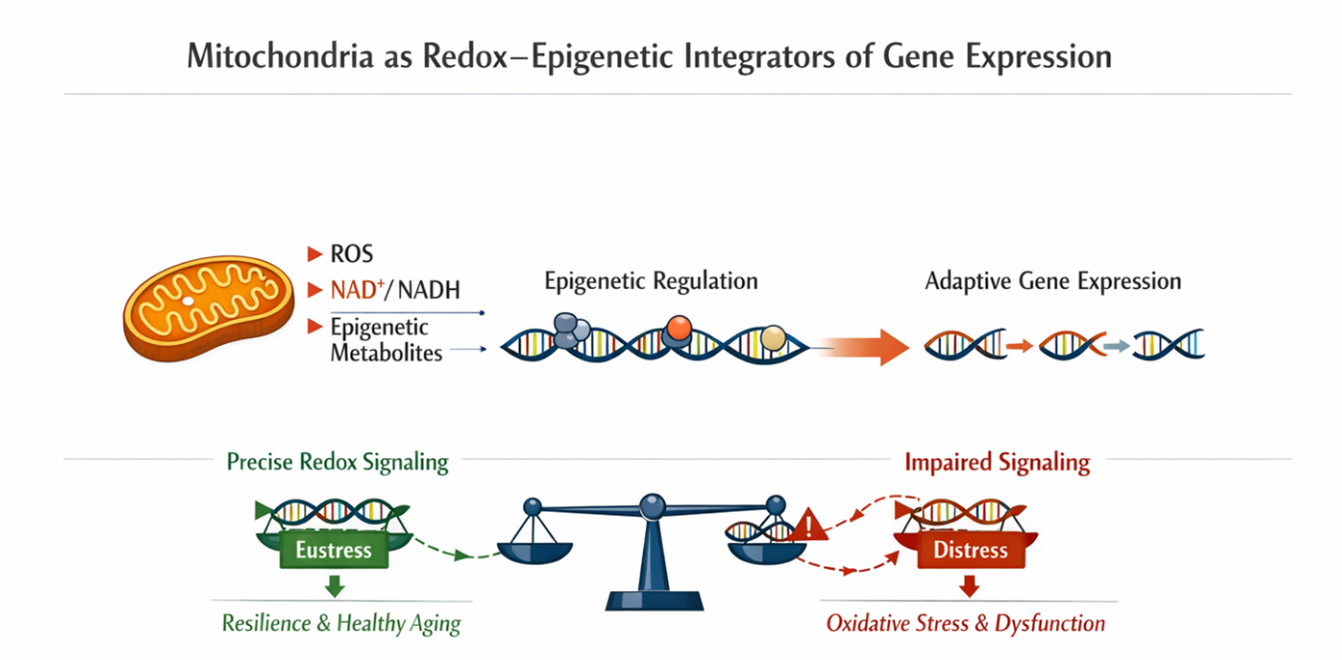
Figure 1. Mitochondria function as redox–epigenetic integrators by translating metabolic and environmental cues into adaptive gene expression programs. Mitochondrial redox outputs, including reactive oxygen species (ROS), NAD+/NADH balance, and epigenetically active metabolites, converge on chromatin regulatory mechanisms to modulate transcriptional responses. When redox signaling is precise and reversible, it promotes epigenetic plasticity, cellular resilience, and healthy aging, reflecting a state of adaptive redox eustress. In contrast, impaired or persistent redox signaling shifts signaling toward oxidative distress, leading to epigenetic dysfunction and loss of adaptive capacity.
|
Mitochondrial redox output |
Context of signaling |
Epigenetic / transcriptional interface |
Functional outcome |
|
NAD+ / NADH balance |
Changes in mitochondrial metabolic flux and redox state |
NAD+-dependent chromatin remodeling, transcriptional control, DNA repair |
Supports transcriptional adaptability and efficient stress resolution |
|
Reactive oxygen species (ROS) |
Localized, reversible redox signaling |
Redox-sensitive transcription factors and chromatin-associated proteins |
Coordinates adaptive gene expression (“eustress”) |
|
Chronic or excessive ROS |
Aging, chronic stress, impaired redox control |
Oxidative modification of epigenetic machinery |
Epigenetic dysfunction and loss of adaptive responsiveness |
|
Redox equivalents and reactive intermediates |
Dynamic physiological contexts (nutrients, exercise, circadian transitions, immune activation) |
Nuclear chromatin regulatory machinery |
Enables context-appropriate transcriptional programs |
|
Overall redox signal fidelity |
Balance between precise vs persistent signaling |
Epigenome as an interpretive and regulatory layer |
Determines epigenetic plasticity, resilience, and healthy aging |
Redox Signaling as Information: The Importance of Fidelity
A critical feature of redox biology is that not all oxidation is equivalent. Adaptive signaling depends on discrete, reversible, and compartmentalized redox events, whereas chronic or excessive oxidation degrades signal fidelity. This distinction has important implications for epigenetic regulation. Because epigenetic systems act as signal interpreters, they require oxidation to behave like information, not damage. When redox events are discrete, reversible, and localized, they enable adaptive gene regulation. However, when oxidation becomes excessive or chronic, epigenetic control degrades, undermining cellular resilience and healthy aging [5,8].
The epigenetic remodeling process is inherently dynamic, involving stimulus-dependent chromatin opening followed by re-establishment of repressive states. Precise redox signals can synchronize this process, facilitating appropriate transcriptional responses and timely resolution. By contrast, persistent redox noise may promote maladaptive chromatin configurations, including sustained stress responses, inflammatory priming, or epigenetic rigidity.
This framework suggests that age-related oxidative changes are not simply the result of accumulating damage but reflect a gradual loss of redox balance. As redox signals become noisier and less controlled, cells struggle to properly coordinate gene expression in response to changing demands. This view also helps explain why broad, non-targeted antioxidant approaches have often produced mixed results, as they may suppress beneficial redox signaling along with harmful oxidative reactions [9].
A Mitochondrial Redox–Epigenetic Axis for Adaptive Gene Expression
A growing body of literature has highlighted an integrated relationship between mitochondrial sensing, redox signaling, and epigenetic regulation. Across diverse physiological contexts, such as nutrient availability, exercise, circadian transitions, and immune activation, mitochondrial metabolic flux and redox state are dynamically modulated, shaping the production of redox equivalents, reactive intermediates, and epigenetically active metabolites.
These outputs converge on nuclear chromatin-regulatory machinery, shaping DNA methylation, histone modification patterns, and transcription factor activity, ultimately enabling adaptive gene expression programs that restore cellular homeostasis. When adaptation is successful, redox signaling resolves, chromatin states remain flexible, and epigenetic plasticity is preserved [4,10].
However, with aging, chronic stress exposure, mitochondrial redox signaling may become dysregulated, either through signal attenuation or persistent activation, when ROS signals are generated at the wrong time or place, or at inappropriate intensity, shifting normally beneficial redox “eustress” into detrimental oxidative “distress”. This impaired communication may reduce epigenetic responsiveness, favoring dysfunctional transcriptional programs that contribute to declining resilience and increased susceptibility to age-associated dysfunction.
Implications for Healthy Aging and Resilience
Epigenetic clocks and other static biomarkers are useful for estimating biological age, but they provide little information about how well cells adapt to stress or change. In contrast, a mitochondrial redox–epigenetic perspective focuses on cellular flexibility: the capacity to shift between functional states, activate appropriate gene expression programs, and efficiently return to baseline [11].
From this perspective, healthy aging may be defined less by the absence of stress responses and more by their precision, reversibility, and integration which are the hallmarks of cellular resilience. Effective responses are those that are appropriately scaled, context-specific, and efficiently resolved, allowing cells to return to functional baseline after challenge. By coordinating metabolic flux, redox signaling, and epigenetic regulation, mitochondria act as redox–epigenetic integrators and may therefore serve as central determinants of biological resilience, supporting adaptive capacity across the lifespan [11,12].
Conclusion
Viewed through this lens, mitochondria emerge as central organizing hubs at the intersection of metabolism, gene regulation, and aging not merely as energy producers or sources of oxidative stress, but as generators of purposeful redox signals. Carefully timed and localized redox signaling enables mitochondria to sense cellular state and encode metabolic and environmental information into chromatin-based gene regulatory programs. Preserving this positive, adaptive redox communication is therefore critical for maintaining epigenetic plasticity which is the capacity to mount appropriate transcriptional responses and to resolve them efficiently. In this way, mitochondrial redox–epigenetic integration provides a unifying framework for understanding biological resilience, positioning healthy aging as the sustained capacity to adapt to and recover from stress.
References
2. Kim KH, Lee CB. Socialized mitochondria: mitonuclear crosstalk in stress. Exp Mol Med. 2024 May;56(5):1033–42.
3. Wu R, Li S, Hudlikar R, Wang L, Shannar A, Peter R, et al. Redox signaling, mitochondrial metabolism, epigenetics and redox active phytochemicals. Free Radic Biol Med. 2022 Feb 1;179:328–36.
4. Xu Y, Jian X, Shi L, Shock LS, Chen L, Chow LT, et al. Mitochondria and Epigenetic Regulation: Bidirectional Crosstalk and Emerging Mitochondria-Targeted Degron Tools. Cells. 2026 Jan 6;15(2):95. doi: 10.3390/cells15020095. Erratum in: Cells. 2026 Mar 17;15(6):530. doi: 10.3390/cells15060530.
5. Castejon-Vega B, Cordero MD, Sanz A. How the Disruption of Mitochondrial Redox Signalling Contributes to Ageing. Antioxidants (Basel). 2023 Mar 29;12(4):831.
6. Covarrubias AJ, Perrone R, Grozio A, Verdin E. NAD+ metabolism and its roles in cellular processes during ageing. Nat Rev Mol Cell Biol. 2021 Feb;22(2):119–41.
7. Bhasin S, Seals D, Migaud M, Musi N, Baur JA. Nicotinamide Adenine Dinucleotide in Aging Biology: Potential Applications and Many Unknowns. Endocr Rev. 2023 Nov 9;44(6):1047–73.
8. Bevere M, Di Cola G, Santangelo C, Grazioli E, Marramiero L, Pignatelli P, et al. Redox-based Disruption of Cellular Hormesis and Promotion of Degenerative Pathways: Perspectives on Aging Processes. J Gerontol A Biol Sci Med Sci. 2022 Nov 21;77(11):2195–206.
9. Cheng YW, Liu J, Finkel T. Mitohormesis. Cell Metab. 2023 Nov 7;35(11):1872–86.
10. Zhu D, Li X, Tian Y. Mitochondrial-to-nuclear communication in aging: an epigenetic perspective. Trends Biochem Sci. 2022 Aug;47(8):645–59.
11. Zhang J, Wang S, Liu B. New Insights into the Genetics and Epigenetics of Aging Plasticity. Genes (Basel). 2023 Jan 27;14(2):329.
12. Castejon-Vega B, Cordero MD, Sanz A. How the Disruption of Mitochondrial Redox Signalling Contributes to Ageing. Antioxidants (Basel). 2023 Mar 29;12(4):831