Commentary
Testosterone plays a crucial role in determining the body composition of male mammals, including humans, due to its effects on muscle and fat mass. Age-related declines in serum testosterone levels in men have been linked to loss of skeletal muscle mass and strength, and physical performance [1-4]. This physical disorder, also known as sarcopenia, is a prevalent condition among the elderly, related to skeletal muscle dysfunction and cell apoptosis. Although the exact mechanisms underlying muscle loss with aging are not fully understood, accumulating evidence postulates that accelerated muscle cell apoptosis may play a central mechanism responsible for impairment of muscle performance [5,6]. Our previous research has shown that testosterone protects against oxidative stress H2O2-induced apoptosis in the C2C12 skeletal muscle cells at multiple levels, encompassing morphological, physiological, and biochemical aspects [7-9], playing the androgen receptor (AR) an active role in these events. Moreover, biochemical and immunological data provided by our laboratory has supported the non-classical localization of the AR in mitochondria and microsomes of C2C12 skeletal muscle cells [10], from where it could be participating in the antiapoptotic effect of testosterone on skeletal muscle [8]. Thus, non-classical localization of AR from where it can exert non-genomic actions could be possible.
Within skeletal muscle, mitochondrial oxidative phosphorylation stands as a primary source of energy, fulfilling essential metabolic requirements and powering physical activity. With advancing age, mitochondria are often reported to diminish in volume and function in muscle, establishing a correlation between the progressive decline in skeletal mitochondrial function and aging, wherein physical strength diminishes disproportionately to muscle mass loss [11,12]. The precise mechanisms through which aging impacts mitochondrial function are not fully understood and continue to be elucidated.
In order to generate fully functional and high-quality organelles, a precise coordination between the nuclear and mitochondrial genomes is imperative to ensure the production of protein products in the correct stoichiometry. Mitochondrial biogenesis needs the expression of numerous genes encoded by both nuclear and mitochondrial genomes [13]. However, given that the protein coding capacity of mitochondrial DNA (mtDNA) is limited to only 13 respiratory subunits, the majority of products required for mitochondrial oxidative functions and biosynthetic capacity must be provided by nuclear genes. Moreover, nuclear genes must play a major role in controlling mitochondrial transcription, translation, and DNA replication. The nuclear respiratory factor-1 (NRF-1) transcription factor has been proposed to play a pivotal role in orchestrating the transcription of both nuclear- and mitochondrial-encoded genes [14]. NRF-1 target genes encompass subunits of the five mitochondrial respiratory chain (MRC) complexes, assembly factors crucial for the respiratory apparatus, components essential for mtDNA transcription and replication, mitochondrial protein import machinery, heme biosynthesis enzymes, and three critical mtDNA transcription factors: mitochondrial transcription factor A (Tfam), mitochondrial transcription factor B types 1 and 2 (TFB1M and TFB2M) [15]. TFAM, TFB1M, and TFB2M transcribe the mitochondrial genome, leading to an increment in mitochondrial-encoded subunits of the MRC [15]. Thus, NRF-1 plays a relevant role in the integration of the nucleus-mitochondria interactions [16].
In this study, we demonstrated that physiological concentrations of testosterone induce the expression of Nrf-1, a phenomenon that triggers an upregulation not only of Tfam but also of TFB2M in skeletal muscle cells. Of relevance, we observed that these effects are reversed in the presence of the AR antagonist, Flutamide. Given that testosterone induces Tfam expression and potentially TFB2M via NRF-1 activation, it is plausible to suggest that testosterone and AR collectively stimulate the mtDNA transcription by upregulating these mitochondrial transcription factors. Consequently, considering that TFAM and TFBs transcribe the mitochondrial genome, testosterone treatment resulted in an increase of mRNA transcription levels of mtDNA-encoded protein subunits comprising the MRC. Specifically, mRNA expression of complex I, III, and IV subunits of the mitochondrial respiratory chain, including ND1, ND4, CytB, Cox1, and Cox2, were significantly increased following testosterone treatment. Notably, the employment of Flutamide was able to almost totally inhibit the effect of the hormone, showing the requirement of the AR for the modulation of these modulatory effects. These findings underscore the significant role of testosterone in modulating mitochondrial gene expression in skeletal muscle. These results strongly suggest that testosterone indirectly regulates mitochondrial gene expression in the skeletal muscle, probably via the activation of the NRF/TFAM-TFBM/mitochondrial genes axis. We propose testosterone's involvement in mitochondrial biogenesis through this primary mechanism of action, modulating mitochondrial gene transcription and contributing to the proper functioning of mitochondrial machinery. These results contribute to a deeper understanding of the involvement of testosterone and its receptor in mitochondrial biogenesis, as a key action of their protective effect against H2O2-induced apoptosis in C2C12 skeletal muscle cells.
H2O2 has been established as a signaling molecule implicated in various cellular processes, including apoptosis [17,18], differentiation [19,20], and proliferation [21]. Notably, H2O2 triggers apoptosis in C2C12 cells in a time-dependent manner. Brief exposures to H2O2 initiate a cellular defense response to avoid apoptosis, whereas prolonged exposure finally leads to programmed cell death. Concurrently, the loss of mitochondrial membrane potential, opening of the mitochondrial permeability transition pore (mPTP), and release of cytochrome c occur, and it is during this phase that the protective influence of testosterone against apoptosis becomes evident [9,10]. This study demonstrates that oxidative stress induced by H2O2 significantly downregulates the mRNA expression of the MRC proteins ND1, ND4, CytB, Cox1, and Cox2, in C2C12 cells compared to the control in C2C12. Furthermore, the expression of the auxiliary factor for promoter recognition, TFB2M, is also diminished in a time-dependent manner following exposure to the apoptotic agent. Thus, it is plausible that the apoptotic agent disrupts mitochondrial integrity and functionality by inhibiting the NRF/TFAM-TFB2M/mitochondrial genes axis, leading finally to apoptosis of skeletal muscle cells. These effects were totally opposite to those obtained with physiological concentrations of testosterone treatment; strengthen the antiapoptotic role of the steroid in skeletal muscle.
The possibility that testosterone could directly influence mitochondrial oxidative phosphorylation gene transcription by way of cognate receptors present in mitochondria, has also been proposed. It has been documented that mtDNA contains response elements for class I and II receptors, to which the AR belongs [22,23]. Indeed, evidence suggests that androgens, mediated by the AR, not only regulate the expression of nuclear genes encoding certain subunits of the MRC, but also modulate the expression of mitochondrial genes encoding subunits of this chain. This regulatory effect might occur directly by binding to androgen response elements present in mtDNA [24,25]. Given that we have previously reported the non-classical localization of AR within the mitochondria of skeletal muscle cells from where the receptor could be mediating the antiapoptotic effects of testosterone, a goal in the present study was to elucidate a potential pathway underlying the observed testosterone-induced preservation and protection of mitochondria. Specifically, our aim was to investigate whether testosterone exerts a direct regulatory influence on the expression of mitochondrial genes involved in maintaining mitochondrial integrity and functionality, thus contributing to the aforementioned preservation and protection of these organelles. To initiate the exploration of this inquiry, we employed computational methodologies to identify specific DNA sequences within the mouse mitochondrial genome that could act as potential androgen response elements (AREs), offering a putative mechanism through which the AR might regulate mitochondrial gene expression. Unlike nuclear genes, which often have multiple promoters, all mitochondrial genes are expressed together from only three promoters encoded in the regulatory D-loop region [26]. These promoters are recognized by the mitochondrial basal transcriptional apparatus, comprising the mitochondrial RNA polymerase (Polrmt) and the mitochondrial transcription factors TFAM and TFB2M [27,28]. In the present investigation, we identified several putative AR binding sites in the murine mitochondrial genome, not only within the regulatory region (D-loop) where the promoters are located, but also at other sites, including within structural genes and adjacent to ribosomal RNA or transfer RNA genes (Figure 1). The presence of transcription factor binding sites (TFBSs) beyond the D-loop region suggests the potential for AR-mediated effects not only during transcription initiation but also at subsequent stages of the mitochondrial transcription process, such as elongation, release and processing. So, these results show the presence of sequence inside the mitochondrial genome, that the AR could recognize, bind, and exert its function as transcription factor by modulating mitochondrial gene transcription.
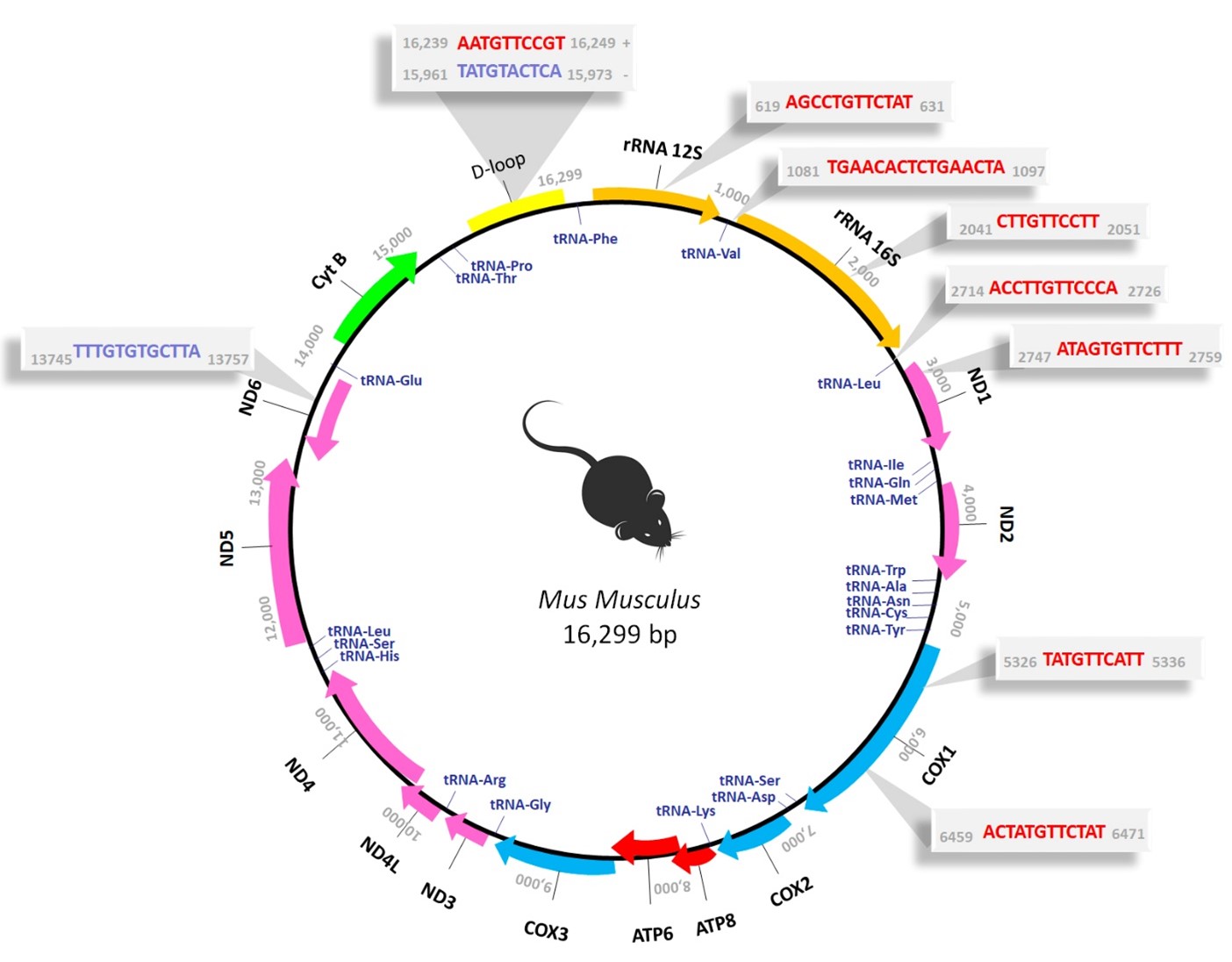
Figure 1: Prediction of putative TFBSs for the AR in the murine mitochondrial genome using computational analysis. Map of Mus Musculus (NC_005089) mitochondrial genome, which comprises 16,299 bp. The mouse mitochondrial DNA (mtDNA) consists of an inner light strand (L-strand) and an outer heavy strand (H-strand), each encoding distinct genes. The H-strand encodes 28 genes, including 2 ribosomal RNAs (rRNAs) (12S and 16S), 14 transfer RNAs (tRNAs) (Phe, Val, Leu, Ile, Met, Trp, Asp, Lys, Gly, Arg, His, Leu, Ser, Thr), and 12 polypeptides (ND1, ND2, ND3, ND4, ND4L, ND5, COX1, COX2, COX3, ATP6, ATP8, CytB). Otherwise, the L-strand encodes only 8 tRNAs (Gln, Ala, Asn, Cys, Tyr, Ser, Glu, Pro) and a singular polypeptide, ND6. Colors identify the different gene categories: rRNAs (orange), tRNAs (blue), and proteins encoding subunits of mitochondrial respiratory chain (MRC) complexes I (pink), III (light blue), IV (green), and V (red). The non-coding region, known as the D-loop, is highlighted in yellow. The gray shaded areas denote the specific sequence binding sites predicted for the AR, with those located on the H-strand depicted in red and those on the L-strand in blue, along with their respective genomic positions (Figure adapted from Pronsato et al. 2020 [29]).
In order to experimentally validate the findings derived from our prior computational analysis, we recently performed the co-immunoprecipitation of the mtDNA from C2C12 cells using an antibody against the AR, followed by sequencing of the precipitated fragments (ChIP-Seq) (unpublished data). This experimental approach enabled us to determine the presence of certain sequences previously identified in silico, thereby confirming the existence of sequences within the mtDNA of C2C12 cells that are recognized and bound by the AR. Consequently, this novel outcome suggests a plausible role for the AR as a mitochondrial transcription factor, potentially exerting a direct regulatory control over mitochondrial gene transcription.
In conclusion, the identification of shared nucleotide sequences in both the nuclear and mitochondrial genomes capable of binding to the AR suggests the possibilities of a coordination of transcription between the two genomes. Consequently, testosterone may, by a direct AR–DNA interaction, stimulate parallel transcription in both nucleus and mitochondria. Moreover, through the induction of nuclear-encoded mitochondrial transcription factors, testosterone could exert a similar effect. Thus, the AR may directly regulate mitochondrial transcription or indirectly lead to the same effect by activating the NRFs/TFAM-TFB2M/mitochondrial genes axis (Figure 2). This study highlights the key role of androgens for mitochondrial gene expression in skeletal muscle and provides an explanation of the antiapoptotic effect of the hormone in aged-skeletal muscle underlying age-related reduction in mitochondrial proteins, function, and quality. Building upon our previous investigations, these findings reinforce the significant contribution of testosterone to the inhibition of various cellular pathways within muscle cells that acting in concert conduce to apoptosis. Clearly, additional studies are necessary to further characterize the precise role of the mitochondrial AR in muscle cell mitochondrial biogenesis and to clarify the signaling mechanisms that mediate the antiapoptotic action of testosterone via the intrinsic apoptotic pathway in skeletal muscle cells, particularly in the context of myopathies associated with hormonal dysregulation.
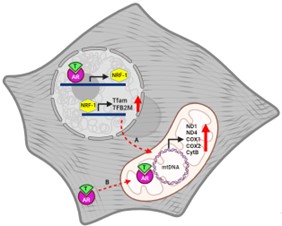
Figure 2: Modulation of mitochondrial gene expression by testosterone in C2C12 cells. A) Indirect Regulation: Testosterone indirectly regulates mitochondrial gene expression in C2C12 cells by activating the AR which leads to the upregulation of Nrf-1, subsequently triggering an increase in the expression of the mitochondrial transcription factors, Tfam and TFB2M. These mitochondrial transcription factors translocate to mitochondria where transcribe the mitochondrial genome, leading to an increment in mitochondrial-encoded subunits of the MRC such as ND1, ND4, CytB, Cox1, and Cox2. B) Direct Regulation: Testosterone may also directly influence mitochondrial oxidative phosphorylation gene transcription by way of cognate receptors present within the mitochondria. These ARs have the capability to recognize and bind specific sequences within the mitochondrial genome, effectively acting as transcription factor, consequently modulating mitochondrial gene transcription.
Testosterone-mediated modulation of mitochondrial gene expression may lead to an increase in mitochondrial biogenesis and function. This can enhance the capacity of skeletal muscle to produce energy through oxidative phosphorylation, improving muscle performance and strength [reviewed in 30]. Testosterone is known to promote muscle protein synthesis and hypertrophy. Testosterone supplementation increases muscle mass in healthy young and old men, healthy hypogonadal men and in other physiological or pathological conditions with low levels of this steroid [31]. Other studies have demonstrated that testosterone induced increase in muscle size is associated with hypertrophy of muscle fibers and significant increases in myonuclear and satellite cell numbers [32-34]. Testosterone's effects on mitochondrial gene expression could potentially help to improve metabolic health by enhancing mitochondrial function and energy metabolism in skeletal muscle, thereby reducing the risk of metabolic diseases. Maximized a mitochondrial biogenesis and function in response to testosterone may lead to improved exercise performance, including greater endurance, strength, and recovery capacity. This can be beneficial for athletes and individuals engaging in regular physical activity.
The loss of muscle mass and strength with aging, that characterized sarcopenia, is a prevalent condition among the elderly and predicts several adverse outcomes. It is a result of reduction in the size and number of muscle fibers [35]. The acceleration of myocyte loss via apoptosis in the elderly might represent the key mechanism responsible for the deterioration of muscle performance [5,6]. Of relevance to this issue, in our laboratory we demonstrated that testosterone protects against oxidative stress-induced apoptosis in skeletal muscle [7-9]. Sarcopenia has been associated with a deficit of sex hormones as the levels of estrogens and androgens decline with aging. Thus, hormone replacement therapies prevent a decline in muscle performance [36,37].
Nevertheless, excessive or prolonged exposure to high testosterone levels can negatively impact mitochondrial function and skeletal muscle metabolism. This can result in a dysregulated mitochondrial gene expression, leading to mitochondrial dysfunction, oxidative stress, and cellular damage, ultimately causing muscle atrophy, decreased performance, even muscle cell apoptosis. The modulation of mitochondrial gene expression by testosterone, may disrupt hormonal balance, especially in individuals with hormonal disorders or undergoing hormone replacement therapy. Such imbalances could affect various physiological processes beyond skeletal muscle, potentially leading to health complications. Misuse of testosterone and other anabolic steroids for performance enhancement or bodybuilding is not uncommon. While manipulating mitochondrial gene expression through exogenous testosterone administration may offer athletic performance benefits, it poses significant health risks and raises ethical concerns.
While modulating mitochondrial gene expression through testosterone in skeletal muscle holds promise for enhancing muscle health, performance, and metabolism, it also presents risks and complexities that require careful control and examination. Further research is essential to gain a deeper understanding of the involved mechanisms and to refine the therapeutic application of testosterone modulation for skeletal muscle function and overall health.
References
2. Ferrando AA, Sheffield-Moore M, Yeckel CW, Gilkison C, Jiang J, Achacosa A, et al. Testosterone administration to older men improves muscle function: molecular and physiological mechanisms. American Journal of Physiology-Endocrinology and Metabolism. 2002 Mar 1;282(3):E601-7.
3. Seidman SN. Androgens and the aging male. Psychopharmacology Bulletin. 2007 Jan 1;40(4):205-18.
4. Krasnoff JB, Basaria S, Pencina MJ, Jasuja GK, Vasan RS, Ulloor J, et al. Free testosterone levels are associated with mobility limitation and physical performance in community-dwelling men: the Framingham Offspring Study. The Journal of Clinical Endocrinology & Metabolism. 2010 Jun 1;95(6):2790-9.
5. Dirks A, Leeuwenburgh C. Apoptosis in skeletal muscle with aging. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2002 Feb 1;282(2):R519-27.
6. Dupont-Versteegden EE. Apoptosis in muscle atrophy: relevance to sarcopenia. Experimental Gerontology. 2005 Jun 1;40(6):473-81.
7. Pronsato L, Ronda AC, Milanesi L, Vasconsuelo A, Boland R. Protective role of 17β-estradiol and testosterone in apoptosis of skeletal muscle. Actual Osteol. 2010;6(2):65-80.
8. Pronsato L, Boland RL, Milanesi LM. Testosterone exerts antiapoptotic effects against H2O2 in C2C12 skeletal muscle cells through the apoptotic intrinsic pathway. J Endocrinol. 2012;212:371-81.
9. Pronsato L, Milanesi L. Effect of testosterone on the regulation of p53 and p66Shc during oxidative stress damage in C2C12 cells. Steroids. 2016 Feb 1;106:41-54.
10. Pronsato L, Boland R, Milanesi L. Non-classical localization of androgen receptor in the C2C12 skeletal muscle cell line. Archives of Biochemistry and Biophysics. 2013 Feb 1;530(1):13-22.
11. Carter CS, Marzetti E, Leeuwenburgh C, Manini T, Foster TC, Groban L, et al. Usefulness of preclinical models for assessing the efficacy of late-life interventions for sarcopenia. Journals of Gerontology Series A: Biomedical Sciences and Medical Sciences. 2012 Jan 1;67(1):17-27.
12. Parise G, De Lisio M. Mitochondrial theory of aging in human age-related sarcopenia. Body Composition and Aging. 2010;37:142-56.
13. Garesse R, Vallejo CG. Animal mitochondrial biogenesis and function: a regulatory cross-talk between two genomes. Gene. 2001 Jan 24;263(1-2):1-16.
14. Evans MJ, Scarpulla RC. Interaction of nuclear factors with multiple sites in the somatic cytochrome c promoter: characterization of upstream NRF-1, ATF, and intron Sp1 recognition sequences. Journal of Biological Chemistry. 1989 Aug 25;264(24):14361-8.
15. Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes & Development. 2004 Feb 15;18(4):357-68.
16. Scarpulla RC. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochimica et biophysica acta (BBA)-Gene Structure and Expression. 2002 Jun 7;1576(1-2):1-14.
17. Singh M, Singh N. Induction of apoptosis by hydrogen peroxide in HPV 16 positive human cervical cancer cells: involvement of mitochondrial pathway. Molecular and Cellular Biochemistry. 2008 Mar;310:57-65.
18. Vasconsuelo AA, Milanesi LM, Boland RL. 17β-Estradiol abrogates apoptosis in murine skeletal muscle cells through estrogen receptors: role of the phosphatidylinositol 3-kinase/Akt pathway. Journal of Endocrinology. 2008;196:385-97.
19. Steinbeck MJ, Kim JK, Trudeau MJ, Hauschka PV, Karnovsky MJ. Involvement of hydrogen peroxide in the differentiation of clonal HD‐11EM cells into osteoclast‐like cells. Journal of Cellular physiology. 1998 Sep;176(3):574-87.
20. Orzechowski A, Grizard J, Gajkowska B, Łokociejewska M, Zaron-Teperek M. Dexamethasone-mediated regulation of death and differentiation of muscle cells. Is hydrogen peroxide involved in the process?. Reproduction Nutrition Development. 2002 May 1;42(3):197-216.
21. Sigaud S, Evelson P, González-Flecha B. H2O2-induced proliferation of primary alveolar epithelial cells is mediated by MAP kinases. Antioxidants & Redox Signaling. 2005 Jan 1;7(1-2):6-13.
22. Demonacos C, Djordjevic-Markovic R, Tsawdaroglou N, Sekeris CE. The mitochondrion as a primary site of action of glucocorticoids: the interaction of the glucocorticoid receptor with mitochondrial DNA sequences showing partial similarity to the nuclear glucocorticoid responsive elements. The Journal of Steroid Biochemistry and Molecular biology. 1995 Oct 1;55(1):43-55.
23. Demonacos CV, Karayanni N, Hatzoglou E, Tsiriyiotis C, Spandidos DA, Sekeris CE. Mitochondrial genes as sites of primary action of steroid hormones. Steroids. 1996 Apr 1;61(4):226-32.
24. Psarra AM, Solakidi S, Sekeris CE. The mitochondrion as a primary site of action of steroid and thyroid hormones: presence and action of steroid and thyroid hormone receptors in mitochondria of animal cells. Molecular and Cellular Endocrinology. 2006 Feb 26;246(1-2):21-33.
25. Usui T, Kajita K, Kajita T, Mori I, Hanamoto T, Ikeda T, et al. Elevated mitochondrial biogenesis in skeletal muscle is associated with testosterone-induced body weight loss in male mice. FEBS letters. 2014 May 21;588(10):1935-41.
26. Montoya J, Christianson T, Levens D, Rabinowitz M, Attardi G. Identification of initiation sites for heavy-strand and light-strand transcription in human mitochondrial DNA. Proceedings of the National Academy of Sciences. 1982 Dec;79(23):7195-9.
27. Falkenberg M, Larsson NG, Gustafsson CM. DNA replication and transcription in mammalian mitochondria. Annu. Rev. Biochem.. 2007 Jul 7;76:679-99.
28. Cotney J, McKay SE, Shadel GS. Elucidation of separate, but collaborative functions of the rRNA methyltransferase-related human mitochondrial transcription factors B1 and B2 in mitochondrial biogenesis reveals new insight into maternally inherited deafness. Human Molecular Genetics. 2009 Jul 15;18(14):2670-82.
29. Pronsato L, Milanesi L, Vasconsuelo A. Testosterone induces up-regulation of mitochondrial gene expression in murine C2C12 skeletal muscle cells accompanied by an increase of nuclear respiratory factor-1 and its downstream effectors. Molecular and Cellular Endocrinology. 2020 Jan 15;500:110631.
30. Dong H, Tsai SY. Mitochondrial properties in skeletal muscle fiber. Cells. 2023 Aug 30;12(17):2183.
31. Bhasin S, Calof OM, Storer TW, Lee ML, Mazer NA, Jasuja R, et al. Drug insight: testosterone and selective androgen receptor modulators as anabolic therapies for chronic illness and aging. Nature clinical practice Endocrinology & metabolism. 2006 Mar;2(3):146-59.
32. Sinha-Hikim I, Artaza J, Woodhouse L, Gonzalez-Cadavid N, Singh AB, Lee MI, et al. Testosterone-induced increase in muscle size in healthy young men is associated with muscle fiber hypertrophy. American Journal of Physiology-Endocrinology and Metabolism. 2002 Jul 1;283:E154–E164.
33. Sinha-Hikim I, Roth SM, Lee MI, Bhasin S. Testosterone induced muscle hypertrophy is associated with an increase in satellite cell number in healthy, young men. American Journal of Physiology-Endocrinology and Metabolism. 2003;285:E197–E205.
34. Sinha-Hikim I, Cornford M, Gaytan H, Lee ML, Bhasin S. Effects of testosterone supplementation on skeletal muscle fiber hypertrophy and satellite cells in community-dwelling older men. The Journal of Clinical Endocrinology & Metabolism. 2006 Aug 1;91(8):3024-33.
35. Lexell J. Ageing and human muscle: observations from Sweden. Canadian Journal of Applied Physiology. 1993 Mar 1;18(1):2-18.
36. Dionne IJ, Kinaman KA, Poehlman ET. Sarcopenia and muscle function during menopause and hormone-replacement therapy. The Journal of Nutrition, Health & Aging. 2000 Jan 1;4(3):156-61.
37. Solomon AM, Bouloux PM. Modifying muscle mass–the endocrine perspective. Journal of Endocrinology. 2006 Nov 1;191(2):349-60.