Clinical Image
A 61-year-old man presented for routine health checkup. The patient was asymptomatic. His medical history was autosomal dominant polycystic kidney disease (ADPKD). Laboratory results and urinalysis were unremarkable. Abdominal computed tomography showed innumerable hepatic cysts of different sizes and diffuse small renal cysts (Figures 1A-1C). Polycystic liver disease (PCLD) is divided into the isolated form and PCLD associated with ADPKD, relevant to genetic defects including PKD1 and PKD2 mutations, which belongs to a family of hepatic ductal plate malformations with variable presence of other components of biliary dysgenesis [1]. PCLD is the most prevalent extrarenal manifestation, which frequently do not compromise liver function but may produce massive hepatomegaly and abdominal discomfort [2]. The liver cysts are not present at birth but develop over time as fluid accumulates, which is commonly modulated by genetic factors, age, pregnancy, and female gender [3]. When complications occur, such as intracystic hemorrhage, infection, cystadenocarcinoma, biliary obstruction, Budd-Chiari syndrome and post-traumatic rupture, surgical intervention is required. Surgical procedure commonly includes cyst aspiration and sclerosis, transcatheter arterial embolization, and fenestration and hepatic resection. In this patient, cyst complications were not identified and he continues routine outpatient follow-up.
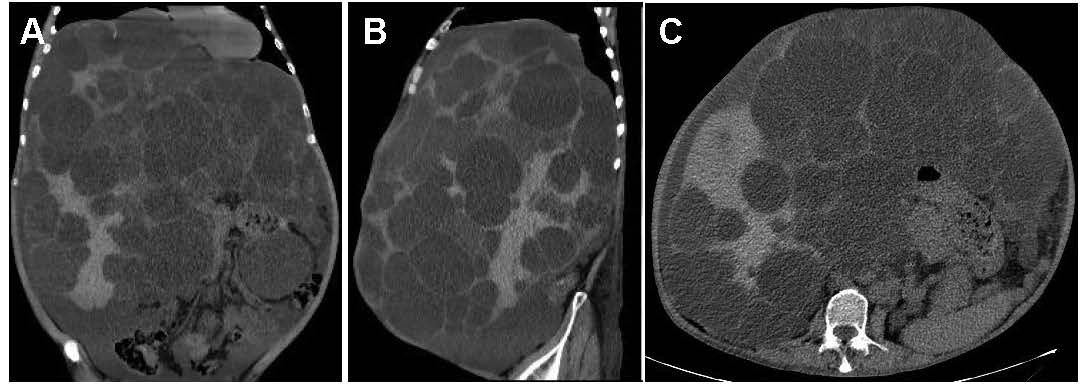
Figure 1. A-C. Abdominal computed tomography reviews.
Conflicts of Interest
The authors have no conflicts of interest to declare.
Ethical Statement
The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. Written informed consent was obtained from the patient for publication of this “GI Image”. Board institutional approval was not required.
Author’s contributions
Collection of data and writing: Han-Sheng Fang.
Manuscript preparation: Han-Sheng Fang.
Final approval of the manuscript: Wei Liu.
Funding
This work was supported by Yichang Medical and Health Research Project (A24-2-008).
References
2. Adu-Gyamfi KO, Kudaravalli P, Yap JEL. Polycystic Liver and Kidney Disease. Clin Gastroenterol Hepatol. 2022 Nov;20(11):A33.
3. van Aerts RMM, van de Laarschot LFM, Banales JM, Drenth JPH. Clinical management of polycystic liver disease. J Hepatol. 2018 Apr;68(4):827–37.