Abstract
GMA301 is a novel antagonistic antibody in clinical development. GMA301 targets human endothelin receptor A (ETA), a proven therapeutic target for pulmonary arterial hypertension (PAH). In preclinical studies, GMA301 demonstrated significant efficacy in lowering pulmonary arterial pressure in both hypoxia-induced and monocrotaline (MCT)-induced PAH monkey models and further attenuated pulmonary arterial and right ventricular hypertrophy in MCT-induced PAH monkeys. GMA301 exhibited an excellent safety profile in cynomolgus monkeys.
GMA301 was also found to be safe and well-tolerated at all intravenous (IV) dosing levels (75 mg, 200 mg, 500 mg, and 1000 mg) in phase 1a clinical studies. No serious adverse events (SAEs) occurred. All of the treatment emergent adverse events (TEAEs) were well tolerated without medical intervention. Its half-life was up to 566 hours, and it is suitable for once-a-month administration. Gmax has initiated US-China multicenter phase 1b clinical studies recently. The monthly dosing regimen, superior safety profile and potentially high efficacy of GMA301 may offer PAH patients a new and effective treatment.
Keywords
Therapeutic antibody, Pulmonary arterial hypertension, Endothelin receptor A, Clinical trials.
Abbreviations
ETA: Endothelin receptor A; GPCR: G Protein-Coupled Receptor; PAH: Pulmonary Arterial Hypertension; ET-1: Endothelin 1; ERA: Endothelin Receptor Antagonist; SAEs: Serious Adverse Events; TEAEs: Treatment Emergent Adverse Events; Mab: Monoclonal Antibody; PK: Pharmacokinetics; MCT: Monocrotaline; RVSP: Right Ventricular Systolic Pressure; PAWT: Pulmonary Arterial Wall Thickness
Pulmonary Arterial Hypertension (PAH)
Pulmonary arterial hypertension (PAH) is a progressive, and life-threatening disease marked by adverse remodeling of small pulmonary arteries and the right ventricle, which causes progressive pulmonary vascular constriction ultimately leading to right ventricular heart failure and death [1-3]. The vascular remodeling is produced mainly by excessive cell proliferation resulting in a gradual narrowing of the vascular luminal cross section and increasing pulmonary vascular resistance (PVR), pulmonary arterial pressure, and severe debilitation [4-6].
Of the 5 categories of pulmonary hypertension described at the 5th World Symposium on Pulmonary Hypertension in 2013, PAH is classified as Group 1 indicating that it is a primary vascular disease. PAH has no clear cause or association (idiopathic) but can be genetically based, drug or toxin-induced or associated with collagen vascular disease, congenital heart disease, human immunodeficiency virus (HIV), portal hypertension or schistosomiasis [7].
ET-1/ETA Axis in PAH
Endothelin-1 (ET-1), a 21-amino-acid long peptide first isolated from the supernatant of cultured endothelial cells, is the most potent vasoconstrictor substance known [8]. Endothelin receptor type A (ETA) and endothelin receptor type B (ETB) belong to the G protein-coupled receptors (GPCRs), assigned to the Gq coupled receptors. The activation of ETA by ET-1 leads to the formation of inositol triphosphate (IP3), which stimulates the contraction of smooth muscle cells by calcium release from the sarcoplasmic reticulum (Figure 1). ETA mediates vasoconstriction and has mitogenic effects on vascular smooth muscle cells, whereas ETB is expressed primarily on vascular endothelial cells and induces endothelial nitric oxide (NO) synthase and acts to clear ET-1 from circulation [9].
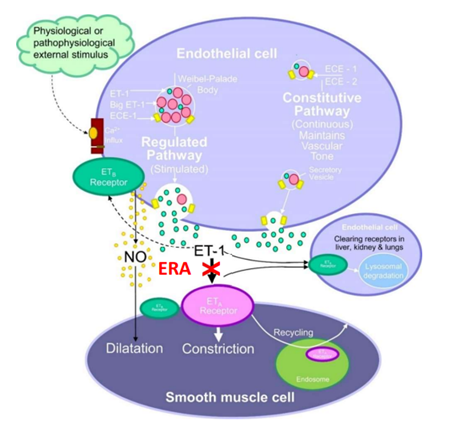
Figure 1: ET-1 Signaling System (ECE: endothelin-converting enzyme).
ET-1 expression level is upregulated in patients with PAH, and ETA is also highly expressed in pulmonary arterial smooth muscle cells [10]. Thus, the ET-1/ETA axis contributes to strong vasoconstriction as well as smooth muscle cell proliferation and is pathophysiologically responsible for PAH [11].
Small Molecular Endothelin Receptor Antagonists (ERAs)
Since 2002, there have been four small molecular drugs targeting the endothelin receptor, including bosentan, sitaxsentan, ambrisentan and macitentan,demonstrating improvements in pulmonary hemodynamics, exercise capacity, functional status, and clinical outcome [12-14]. However, adverse effects included liver transaminase elevation, peripheral edema, anemia, and gastrointestinal reactions. Pfizer withdrew sitaxsentan from the market in 2010 due to several reports of fatal liver injury in PAH patients [15].
Bosentan and macitentan are dual endothelin receptor antagonists [16]. Bosentan is associated with abnormal liver function as a common adverse effect in 10% of PAH patients, while macitentan is associated with a higher incidence of anemia. Ambrisentan is a relatively selective ETA blocker and has demonstrated similar efficacy over bosentan and macitentan [17,18]. Theoretically, selective small molecular antagonists of ETA should be more effective than nonselective small molecular antagonists, given the role played by ETB in both vasodilation and ET-1 clearance [19].
GMA301 as Antibody Therapeutics for PAH
Current therapeutic approaches for PAH commonly include the use of prostacyclins, endothelin pathway antagonists or nitric oxide (NO) pathway modulators. These agents are non-specific and suffer from several important shortcomings, including shorter half-lives, higher dose and frequency requirements, several dosage-related systemic side effects, etc. Hence, there is a critical need to discover new agents that offer both improved therapeutic efficacy as well as a reduction in administration frequency. Our ERA program is aimed at developing more selective therapies for PAH by focusing on novel molecules that act on conventional pathways with improved characteristics.
In addition to having high selectivity for a targeted protein, monoclonal antibodies (MAbs) can also exhibit less off-target actions. Compared to small molecules, monoclonal antibodies (MAbs) have a very long plasma half-life, leading to a prolonged time course of action which can improve patient compliance. Due to their favorable pharmacokinetics (PK), pharmacodynamics (PD) and toxicity profiles, MAbs are seen as more efficacious therapeutics compared to small molecular drugs [20-22].
By applying our GPCR MAb technology platform, we generated an antagonistic MAb GMA301, specifically blocking the interaction between ET-1/ETa (Figure 1) with favorable pharmacokinetics/pharmacodynamics and toxicity profiles [23]. GMA301 was found to have no side effects in liver toxicity or edema in the preclinical studies with cynomolgus monkeys.
Pre-Clinical Study of GMA301
In binding human ETA, GMA301 had an equilibrium dissociation constant (Kd) value of 8.7 nM. The in vitro functional activity of GMA301 was studied in a cell-based calcium influx assay with a half maximal inhibitory concentration (IC50) value of 37.9 nM.
Acute and chronic toxicity studies were also pursued with a no-observed-adverse-effect level (NOAEL) of 750 mg/kg for single dose toxicity and a NOAEL of 250 mg/kg for 4-week toxicity. The 250 mg/kg dosage is 50 times higher than the intended human clinical dose. Even with the high dosage level, GMA301 has a clean safety profile with no abnormalities observed in body weight, blood pressure, food consumption, ECG, and clinical pathology tests.
GMA301 does not cross-react with mouse or rat ETA. The hypoxia-induced PAH monkey model, established by acute exposure to hypoxia, was utilized for efficacy assessment, and GMA301 reduced pulmonary arterial pressure for up to 168 hours. The MCT-induced monkey PAH model was also established to study the pathologic changes before and after treatment with GMA301. In comparison with the PAH model group, the left ventricular hypertrophy index (Fulton’s Index), pulmonary arterial wall thickness (PAWT) and right ventricular systolic pressure (RVSP) of the treatment group were decreased significantly (P-value <0.001) by as much as 38.7%, 49.8% and 54.0%, respectively, after intravenous injection of GMA301 at the 15 mg/kg dose twice per week for 6 weeks (Table 1).
|
Treatments |
Fulton’s Index (%) |
PAWT (%) |
RVSP (mmHg) |
|
Sham |
16.5 ± 2.8 |
9.80 ± 0.32 |
17.0 ± 1.2 |
|
MCT Model |
32.3 ± 1.6### |
23.42 ± 0.82### |
42.0 ± 0.8### |
|
Ambrisentan, 1 mg/kg |
23.1 ± 1.2*** |
14.36 ± 1.05*** |
26.3 ± 2.1*** |
|
GMA301, 1.5 mg/kg |
29.0 ± 1.9## |
20.50 ± 1.25### |
36.3 ± 3.6### |
|
GMA301, 5 mg/kg |
22.3 ± 1.2*** |
15.52 ± 0.93*** |
22.8 ± 1.8*** |
|
GMA301, 15 mg/kg |
19.8 ± 0.6*** |
11.76 ± 0.98*** |
19.3 ± 0.5*** |
Furthermore, GMA301 at the 15 mg/kg dose, equivalent to a human dosage level of 300 mg, was significantly more efficacious than ambrisentan on the right ventricular systolic pressure (RVSP) [23]. At this dose level in PAH cynomolgus monkeys, GMA301 suppressed the rise of pulmonary pressure completely. Notably, as derived from a repeated-dose toxicity study, therapeutic dosage levels can be further increased while maintaining subjects safely.
Clinical Phase Ia Study of GMA301 in Australia
A single-center, double-blind, placebo-controlled, dose escalation study to assess the safety, tolerability, and PK of GMA301 in healthy subjects was conducted in Australia. The study population featured four sequential dosing cohorts, each with 6 subjects receiving GMA301 and 2 subjects receiving a placebo (total of 32 subjects). GMA301 given as single escalating IV injections at 75 mg, 200 mg, 500 mg, and 1000 mg was found to be safe and well-tolerated. No serious adverse events (SAEs) and deaths occurred, and no treatment emergent adverse events (TEAEs) resulted in study discontinuation. No notable findings were observed in laboratory results, vital signs, 12-lead ECG, immunogenicity, and ET-1 levels. The most frequent TEAEs across the 4 groups were headache (39.4%), nausea (12.1%) and pharyngitis (12.1%); however, all of these TEAEs were well tolerated without the need of medical intervention.
Pharmacokinetic results (Figure 2 and Table 2) were obtained from the same study that was conducted in Australia. All individuals in the study exhibited quantifiable plasma GMA301 concentrations at the first timepoint of 4 hours. Most individuals appeared to exhibit mono-exponential PK, with a slow decline in plasma concentrations after reaching maximum observed serum concentration (Cmax). Terminal half-life was long with average values ranging from 503.75 hours to 565.63 hours, thus demonstrating that GMA301 may serve as a once-a-month treatment in PAH patients.
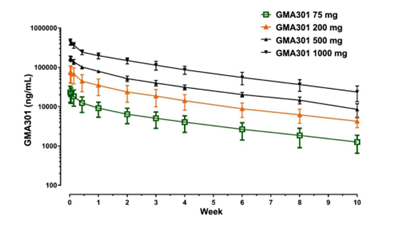
Figure 2: Clinical Pharmacokinetic Data.
|
PK Parameters |
75 mg (N=6) |
200 mg (N=6) |
500 mg (N=6) |
1000 mg (N=6) |
|
T1/2 (h) |
498.4 ± 107.1 |
565.3 ± 60.8 |
536.7 ± 136.6 |
517.7 ± 64.0 |
|
Tmax (h) |
4.0 ± 0.0 |
8.0 ± 8.0 |
5.3 ± 1.9 |
4.7 ± 1.5 |
|
Cmax (mg/mL) |
26.9 ± 4.9 |
89.9 ± 17.3 |
171.8 ± 22.2 |
454.3 ± 64.5 |
On-going Clinical Phase Ib Study of GMA301 in China and USA
This is a phase 1b randomized, placebo-controlled, double-blind, dose escalation study to assess the safety, efficacy and PK of GMA301 in PAH subjects with World Health Organization (WHO) functional class II to III who are taking approved targeted therapies other than endothelin receptor antagonists (ERAs). Treatments will include 3 sequential dose-ascending cohorts, each with 9 subjects receiving GMA301 injections and 3 subjects receiving GMA301 matching placebo injections (total of 36 subjects; in the ratio of 3:1).
The selection of GMA301 doses and dosing frequency are based on the available clinical safety, tolerability and PK data obtained from the healthy human subjects of the Phase Ia study in Australia. The dose level of 1000 mg GMA301 observed in the previous study has been proven safe in human subjects. Patients in the ascending dose cohorts who are assigned to active GMA301 will be administered with 300, 600, or 1000 mg GMA301 every 4 weeks (Q4W) based on their assigned cohort. The study will be carried out at clinical sites in China and the US.
Phase Ib clinical trials will test GMA301 for the first time in a small group of 36 patients over the course of 12 weeks. The goals are to assess the safety of the drug or treatment, find a safe dosage range, identify any side effects, and determine drug efficacy in PAH patients, including the ability to better control pulmonary pressure as well as prevent further remodeling of pulmonary arteries and the right heart ventricle, all of which lead to better quality of life and increased survival rates.
Product Development of GMA301
The Phase Ib study in USA and China will obtain further safety and tolerability data when GMA301 is administered as multiple doses (300, 600 and 1000 mg) in subjects with PAH. Significant drug efficacy may be expected at the 600 mg dose level without SAEs. The intended therapeutic dose will be further evaluated in the following phase II trial and pivotal phase III trial with a larger number of patients, to confirm and compare the safety and effectiveness of GMA301 against the standard treatments.
The safety profile that we have observed so far with the drug candidate GMA301 is very encouraging. GMA301 may allow some PAH patients to achieve meaningful clinical benefits. Additionally, receiving orphan drug designation from the FDA for PAH provides a special opportunity to serve a group of patients who have a poor prognosis with currently available treatments. Due to its superior safety and convenient monthly dosing frequency, GMA301 could additionally become a significant therapeutic for children with PAH.
Acknowledgements
The team of Gmax Biopharm is fully acknowledged for the work described in this paper.
References
2. Barst RJ, McGoon M, Torbicki A, Sitbon O, Krowka MJ, Olschewski H, et al. Diagnosis and differential assessment of pulmonary arterial hypertension. Journal of the American College of Cardiology. 2004 Jun 16;43(12 Supplement): S40-7.
3. Lai YC, Potoka KC, Champion HC, Mora AL, Gladwin MT. Pulmonary arterial hypertension: the clinical syndrome. Circulation Research. 2014 Jun 20;115(1):115-30.
4. Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. Journal of the American College of Cardiology. 2004 Jun 16;43(12 Supplement): S13-24.
5. McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation task force on expert consensus documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation. 2009 Apr 28;119(16):2250-94.
6. Lau EM, Giannoulatou E, Celermajer DS, Humbert M. Epidemiology and treatment of pulmonary arterial hypertension. Nature Reviews Cardiology. 2017 Oct;14(10):603.
7. Humbert M, Lau EM, Montani D, Jaïs X, Sitbon O, Simonneau G, et al. Advances in therapeutic interventions for patients with pulmonary arterial hypertension. Circulation. 2014 Dec 9;130(24):2189-208.
8. Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988 Mar;332(6163):411-5.
9. Davenport AP, Hyndman KA, Dhaun N, Southan C, Kohan DE, Pollock JS, et al. Pharmacological Reviews. 2016 Apr 1;68(2):357-418.
10. Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. New England Journal of Medicine. 1993 Jun 17;328(24):1732-9.
11. Barton M, Yanagisawa M. Endothelin: 20 years from discovery to therapy. Canadian journal of Physiology and Pharmacology. 2008 Aug;86(8):485-98.
12. Rubin LJ, Badesch DB, Barst RJ, Galiè N, Black CM, Keogh A, et al. Therapy for pulmonary arterial hypertension. New England Journal of Medicine. 2002 Mar 21;346(12):896-903.
13. Galiè N, Olschewski H, Oudiz RJ, Torres F, Frost A, Ghofrani HA, et al. Pulmonary Arterial Hypertension, Randomized, Double-Blind, Placebo-Controlled, Multicenter, Efficacy Studies (ARIES) Group. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation. 2008 Jun 10;117(23):3010-9.
14. Iglarz M, Binkert C, Morrison K, Fischli W, Gatfield J, Treiber A, et al. Pharmacology of macitentan, an orally active tissue-targeting dual endothelin receptor antagonist. Journal of Pharmacology and Experimental Therapeutics. 2008 Dec 1;327(3):736-45.
15. Barst RJ, Langleben D, Badesch D, Frost A, Lawrence EC, Shapiro S, et al. Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentan. Journal of the American College of Cardiology. 2006 May 16;47(10):2049-56.
16. Blok IM, van Riel AC, van Dijk AP, Mulder BJ, Bouma BJ. From bosentan to macitentan for pulmonary arterial hypertension and adult congenital heart disease: further improvement? International Journal of Cardiology. 2017 Jan 15; 227:51-2.
17. Pharmacology/Toxicology Review and Evaluation, FDA Drug Approval Package: Letairis (Ambrisentan) NDA #022081, 2007.
18. Casserly B, Klinger JR. Ambrisentan for the treatment of pulmonary arterial hypertension. Drug Des Devel Ther. 2009 Feb 6;2:265-80.
19. Seferian A, Simonneau G. Therapies for pulmonary arterial hypertension: where are we today, where do we go tomorrow? European Respiratory Review. 2013 Sep 1;22(129):217-26
20. Zhao L, Shang EY, Sahajwalla CG. Application of pharmacokinetics–pharmacodynamics/clinical response modeling and simulation for biologics drug development. Journal of Pharmaceutical Sciences. 2012 Dec 1;101(12):4367-82.
21. Deng R, Jin F, Prabhu S, Iyer S. Monoclonal antibodies: what are the pharmacokinetic and pharmacodynamic considerations for drug development? Expert Opinion on Drug Metabolism & Toxicology. 2012 Feb 1;8(2):141-60.
22. Kamath AV. Translational pharmacokinetics and pharmacodynamics of monoclonal antibodies. Drug Discovery Today: Technologies. 2016 Sep 1;21:75-83.
23. Zhang C, Wang X, Zhang H, Yao C, Pan H, Guo Y, Fan K, et al. Therapeutic Monoclonal Antibody Antagonizing Endothelin Receptor A for Pulmonary Arterial Hypertension. Journal of Pharmacology and Experimental Therapeutics. 2019 Jul 1;370(1):54-61.