Abstract
Activated phosphoinositide-3-kinase delta syndrome (APDS) is an autosomal dominant immune dysregulation due to the gain of function mutation of phosphoinositide-3-kinase Delta (PI3Kδ). Patients with APDS typically present with recurrent infections, lymphoproliferation, autoimmunity, and increased risk for malignancies. Herein, we report a case of APDS who presents as an infant with thrombocytopenia at the age of 4 months. He was found to have anti-platelet antibodies. Therefore, he was diagnosed with Immune thrombocytopenic purpura (ITP) and was treated successfully with high dose intravenous immune globulin (IVIG). The patient’s immunology workup was notable for elevated IgM level, T-cell and NK cell lymphopenia with a decreased percentage of naïve CD4 and CD8 cells and a marked increase in transitional B cells, a hallmark of APDS. The genetic test revealed a pathogenic variant in the PIK3CD gene (c.3061G>A p.Glu1021Lys) consistent with APDS diagnosis.
Keywords
Activated phosphoinositide-3-kinase delta syndrome, PIK3CD gene, Autoimmune thrombocytopenia
Abbreviations
APDS: Activated Phosphoinositide-3-kinase Delta Syndrome; PI3Kδ: Phosphoinositide3-kinase Delta; ITP: Immune Thrombocytopenic Purpura; IVIG: Intravenous Immune Globulin; HSV: Herpes Simplex Virus; VZV: Varicella-Zoster Virus; CMV: Cytomegalovirus; EBV: Epstein Barr Virus; SLE: Systemic Lupus Erythematosus; ANCA: Antineutrophil Cytoplasmic Antibody; TRECs: T cell Receptor Excision Circles; ARPC1B: Actin-related protein 2/3 complex subunit 1B; LRBA: Lipopolysaccharide-Responsive and Beigelike Anchor protein; CTLA-4: Cytotoxic T-lymphocyte-Associated Protein 4
Introduction
Activated phosphoinositide 3-kinase delta syndrome (APDS) is a rare syndrome of immune dysregulation that is inherited in an autosomal dominant fashion and results in varying severity of the clinical phenotype [1,2]. APDS is a result of a gain-of-function mutation in the gene PIK3CD, which codes for a catalytic subunit of the PI3K-Delta kinase. Over-activation of this kinase leads to mTOR activation, promotion of cell growth and protein synthesis, and inhibition of apoptosis, leading to lymphoproliferation, recurrent infections, and autoimmunity [1,2]. The lymphoproliferation features can range from nonneoplastic lymphoproliferation resulting in chronic lymphadenopathy and hepatosplenomegaly to lymphoma and leukemia [1,3-6]. Recurrent infections are very common and likely due to the defective T and B cell receptor signaling and decreased memory cell formation [2]. The most common reported infection is upper and lower respiratory tract infections which can lead to bronchiectasis in up to 75% of the cases [1,7,8]. Also, many of these patients have persistent or recurrent herpes virus infections such as HSV, VZV, EBV, and CMV, which is consistent with impaired T cell function [2]. Lastly, APDS is associated with an increased risk of autoimmune diseases such as autoimmune cytopenia, thyroid disease, SLE and ANCA vasculitis [1,4,9]. Herein, we present a case of APDS who presents as an infant with Autoimmune Thrombocytopenia (ITP).
Case Presentation
A 4-month-old boy, was born post-term at 42 weeks via emergency C-section due to non-reassuring fetal heart tones. Meconium aspiration was noted at birth and the patient was found to have petechiae and thrombocytopenia. He needed a three-week NICU stay for respiratory support and required a platelet transfusion during admission. His newborn screen for severe combined immunodeficiency by quantifying T cell receptor excision circles (TRECs) was negative. At two months old, the patient required brief admission for parainfluenza and rhinovirus respiratory infection. The platelet count was normal on admission. A month later, he presented with a cough and recurrence of petechiae. He was found to have pneumonia and thrombocytopenia. He was found to have anti-platelet antibodies. Therefore, he was diagnosed with Immune thrombocytopenic purpura (ITP) and was treated successfully with high dose intravenous immune globulin (IVIG) (Figure 1).
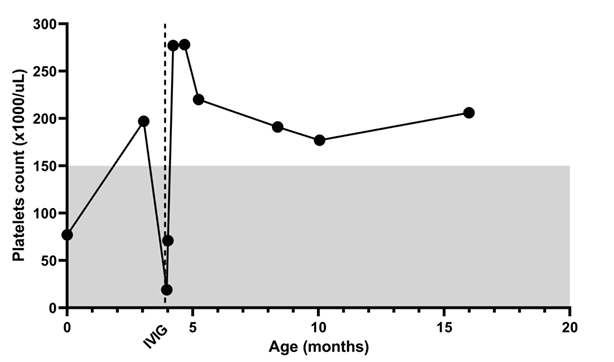
Figure 1: Platelet count during the disease course.
The differential diagnosis of infantile thrombocytopenia can be divided into two major groups: lack of platelet production and increased consumption. Lack of production could be due to: 1- inherited thrombocytopenias (such as Amegakaryocytic thrombocytopenias, thrombocytopenia and absent radii, Wiskott-Aldrich syndrome, or ARPC1B deficiency) [10]; or 2- congenital infection (such as congenital CMV, or congenital rubella). These diseases typically present at birth and have a chronic course, especially in the inherited thrombocytopenias. Increased platelet consumption can be secondary to immune-mediated processes (such as neonatal alloimmune thrombocytopenia and ITP) or coagulopathy. In the case we are reporting here, the initial thrombocytopenia is likely due to meconium aspiration and possible infection. The patient's platelet count recovered completely at the age of three months (Figure 1) which makes hereditary thrombocytopenias unlikely. A month later, he had more severe thrombocytopenia, which is likely to be unrelated to the initial thrombocytopenia. He didn't have lab findings to support the diagnosis of disseminated intravascular coagulation or hemophagocytic lymphohistiocytosis. Therefore, the cause of thrombocytopenia at four months is most likely immune-mediated, which is supported by the presence of the anti-platelet antibodies. The early onset of the autoimmune raised suspicion for primary immune dysregulation disorders such as RAG1, RAG2, TNFRSF13B, CTLA4, LRBA, PIK3CD, and FOXP3 genetic variant.
He had a thorough immunology workup because of his history of early-onset ITP and pneumonia. His immunoglobulins level revealed elevated IgM at 237 mg/dl. Flow cytometry was notable for T-cell and NK cell lymphopenia with low levels of naïve CD4 and CD8 cells (Table 1). B cell subset showed a marked increase in transitional B cells. Given abnormal immunology workup, primary immunodeficiency genetic panel was obtained and revealed a pathogenic variant in the PIK3CD gene (c.3061G>A p.Glu1021Lys.) consistent with the diagnosis of activated phosphoinositide 3-kinase delta syndrome.
|
|
Reference range |
Results |
|
Complete Blood Count |
|
|
|
White Blood Cells |
5.0 - 19.5 thou/uL |
10.07 |
|
Red Blood Cell Count |
3.10 - 4.50 mil/uL |
3.28 |
|
Hemoglobin |
9.0 - 14.0 g/dL |
9.3 |
|
Hematocrit |
29 - 41 % |
28.3 (L) |
|
MCH |
25.0 - 35.0 pg |
28.5 |
|
MCHC |
30.0 - 36.0 g/dL |
33.0 |
|
MCV |
74.0 - 108.0 fL |
86.5 |
|
Platelets |
150 - 450 thou/uL |
25 (L) |
|
Mean Platelet Volume |
7.4 - 10.4 fL |
9.1 |
|
RBC Dist Width |
12.5 - 16.0 % |
14.7 |
|
Monocytes |
3 - 10 % |
5.0 |
|
Lymphocytes |
44 - 74 % |
67.6 |
|
Neutrophils |
14 - 34 % |
23.9 |
|
Eosinophils |
0 - 6 % |
1.5 |
|
Basophils |
0 - 2 % |
0.5 |
|
Immunoglobulin levels |
|
|
|
Immunoglobulin A (IGA) |
4.6 - 46 mg/dL |
27.8 |
|
Immunoglobulin E (IGE) |
<9.2 KU/L |
3.36 |
|
Immunoglobulin G (IGG) |
169 - 558 mg/dL |
467 |
|
Immunoglobulin M (IGM) |
23 - 85 mg/dL |
237 (H) |
|
T, B, NK cell flowcytometry |
|
|
|
CD3%(pan T-cells) |
55 - 79 % |
25 (L) |
|
CD3 absolute count |
2,533 - 6,778 /mm3 |
1,631 (L) |
|
CD3+CD4+% (T-helper cell) |
35 - 62 % |
22 (L) |
|
CD3+CD4+ absolute count |
1,392 - 5,210 /mm3 |
1,383 (L) |
|
CD3+CD8+% (T- cytotoxic cells) |
14 - 30 % |
4 (L) |
|
CD3+CD8+ absolute count |
652 - 2,449 /mm3 |
231 (L) |
|
CD4/CD8 ratio |
1.29 - 4.14 |
5.99 (H) |
|
CD19% (pan B-cells) |
14 - 39 % |
72 (H) |
|
CD19 absolute count |
745 - 3,499 /mm3 |
4,645 (H) |
|
CD3-CD16CD56+% (NK cells) |
3 - 14 % |
2 (L) |
|
CD3-CD16CD56+ absolute count. |
194 - 994 /mm3 |
124 (L) |
|
B cell subsets |
|
|
|
Isotype-switched memory B cell |
0.01 - 4.71 % B Cells |
0.56 |
|
Isotype-unswitched memory B cell |
0.30 - 8.17 % B Cells |
8.14 |
|
Naive B cells |
86.74 - 98.34 % B Cells |
86.93 |
|
Plasmablast |
0.05 - 3.66 % B Cells |
0.22 |
|
Transitional B cell (CD21-CD10+) |
0.00 - 0.21 % B Cells |
51.90 (H) |
|
T cell subsets |
|
|
|
|
|
|
|
|
|
|
|
CD8 CENTRAL MEMORY CELLS |
0.11 - 2.56 % of CD8 T Cells |
1.45 |
|
CD8 Effector Memory Cells |
0.37 - 10.32 % of CD8 T Cells |
31.76 (H) |
|
CD8 Naïve T Cells |
47.10 - 91.30 % of CD8 T Cells |
22.92 (L) |
|
CD4 Central Memory Cells |
2.73 - 14.50 % of CD4 T Cells |
32.82 (H) |
|
CD4 Effector Memory Cells |
0.43 - 5.53 % of CD4 T Cells |
4.99 |
|
CD4 Naïve T Cells |
61.90 - 89.05 % of CD4 T Cells |
27.76 (L) |
Discussion
This case highlights the importance of considering primary immune dysregulation in patients who present with early onset of autoimmunity, especially in patients with a history of severe infection. To date, there are more than 400 distinct genetic disorders affecting the immune system, and many of them can present with autoimmunity as well as recurrent infection [11]. Identification of the genetic variant (mutation) can provide insight into the disease's pathophysiology and allow the opportunity for targeted therapies. For example, LRBA (Lipopolysaccharide-responsive and beige-like anchor protein) deficiency decreases the expression of CTLA-4 (cytotoxic T-lymphocyte-associated protein 4) on the T regulatory cells which is an essential protein to inhibit the immune response. These patients respond well to abatacept which is a CTLA4 fusion protein [12,13]. This patient had a pathogenic variant in the PIK3CD gene (c.3061G>A p.Glu1021Lys.), which has been reported in 17 patients with APDS [14]. Because this variant leads to over-activation of phosphoinositide 3-kinase delta, and therefore activation of the mTOR pathway, mTOR inhibitors such as rapamycin or sirolimus can be good treatments for autoimmunity or lymphoproliferation. Currently, there are ongoing clinical trials for the use of selective PI3KD inhibitors to treat patients with APDS. Finally, hematopoietic stem cell transplantation can offer a curative approach to this disease but also can carry the risk of causing graft-versus-host disease [15]. Establishing the early molecular diagnosis is important to provide proper family counseling and discuss the risk of future complications. For example, APDS is an autosomal dominant disease; therefore, there is a 50% chance that future offspring will have the same disease. Given the high risk for recurrent pneumonia and bronchiectasis, early treatment with IgG replacement therapy for APDS patients with hypogammaglobulinemia or poor vaccine response is advisable [1,7,14]. Also, APDS patients are at a higher risk of lymphoma; therefore, low threshold biopsy for suspicion of the lymph node is recommended [1].
Conclusions
Immunology workup should be considered in patients with early onset of autoimmunity, especially in patients with a history of severe infection. Early genetic testing can provide insight into the disease's pathophysiology and allow the opportunity for targeted therapies. Increased transitional B cell count can be an important clue for the diagnosis of activated phosphoinositide-3-kinase delta syndrome.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this article
Funding statement
The authors do not have grant or funding for this publication
Acknowledgments
The authors like to acknowledge the parents' participation in the case report and the clinical immunology lab at Ann & Robert H. Lurie Children's Hospital of Chicago for running the immunology labs.
References
2. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U et al. Dominant-activating germline mutations in the gene encoding the PI (3) K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nature Immunology. 2014 Jan;15(1):88-97.
3. Yin Z, Tian X, Zou R, He X, Chen K, Zhu C. Case Report: First Occurrence of Plasmablastic Lymphoma in Activated Phosphoinositide 3-Kinase δ Syndrome. Frontiers in Immunology. 2021;12:813261.
4. Conti F, Catelli A, Cifaldi C, Leonardi L, Mulè R, Fusconi M, et al . Case Report: Hodgkin Lymphoma and Refractory Systemic Lupus Erythematosus Unveil Activated Phosphoinositide 3-Kinase-δ Syndrome 2 in an Adult Patient. Frontiers in Pediatrics. 2021;9: 702546.
5. Kracker S, Curtis J, Ibrahim MA, Sediva A, Salisbury J, Campr V et al. Occurrence of B-cell lymphomas in patients with activated phosphoinositide 3-kinase δ syndrome. Journal of Allergy and Clinical Immunology. 2014 Jul 1;134(1):233-6.
6. Pham MN, Cunningham-Rundles C. Evaluation of lymphoproliferative disease and increased risk of lymphoma in activated phosphoinositide 3 kinase delta syndrome: a case report with discussion. Frontiers in Pediatrics. 2018 Dec 18;6:402.
7. Fekrvand S, Delavari S, Chavoshzadeh Z, Sherkat R, Mahdaviani SA, Sadeghi Shabestari M, et al. The first iranian cohort of pediatric patients with activated phosphoinositide 3-kinase-δ (PI3Kδ) syndrome (APDS). Immunological Investigations. 2022 Apr 3;51(3):644-59.
8. Condliffe AM, Chandra A. Respiratory manifestations of the activated phosphoinositide 3-kinase delta syndrome. Frontiers in Immunology. 2018 Mar 5;9:338.
9. Zhang X, Wang J, Zhu K, Jin Y, Fu H, Mao J. Activated phosphoinositide 3-kinase delta syndrome misdiagnosed as anti-neutrophil cytoplasmic antibody-associated vasculitis: a case report. Journal of International Medical Research. 2021 May;49(5):03000605211013222.
10. Cines DB, Bussel JB, McMillan RB, Zehnder JL. Congenital and acquired thrombocytopenia. ASH Education Program Book. 2004;2004(1):390-406.
11. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al. Human inborn errors of immunity: 2022 update on the classification from the international union of immunological societies expert committee. Journal of Clinical Immunology. 2022 Jun 24:1-35.
12. Tesch VK, Abolhassani H, Shadur B, Zobel J, Mareika Y, Sharapova S, et al. Long-term outcome of LRBA deficiency in 76 patients after various treatment modalities as evaluated by the immune deficiency and dysregulation activity (IDDA) score. Journal of Allergy and Clinical Immunology. 2020 May 1;145(5):1452-63.
13. Bukhari A, Ortega-Treviño MF, Padem N, Khojah AM. Hydroxychloroquine Does not Affect CTLA-4 Expression in LRBA Deficiency: Case Report from Siblings with LRBA Deficiency. Journal of Pediatrics and Congenital Disorders. 2020;6(1):1-4.
14. Angulo I, Vadas O, Garçon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Science. 2013 Nov 15;342(6160):866-71.
15. Notarangelo LD. Hematopoietic stem cell transplantation for activated phosphoinositide 3-kinase δ syndrome: Who, when, and how?. Journal of Allergy and Clinical Immunology. 2019 Jan 1;143(1):91-3.