Abstract
N,N-dimethyltryptamine (DMT) is an alkaloid structurally similar to serotonin that acts on serotonergic receptors. This study aimed to evaluate the effects of DMT on peripheral nociception and explore its possible mechanisms of action. Male Wistar rats underwent a mechanical paw pressure test, with hyperalgesia induced by intraplantar injection of prostaglandin E2 (PGE2; 2 µg/paw). Pretreatment with ketanserin, ondansetron, isamoltan, naloxone, AM251, AM630, L-NOArg, or glibenclamide was administered 35 minutes before testing. Nociceptive thresholds were measured 180 minutes after PGE2 injection. DMT (2.5, 10, and 40 μg/paw) showed a dose-dependent peripheral antinociceptive effect against PGE2-induced hyperalgesia, with the highest dose not affecting the contralateral paw. The antinociceptive effect of DMT was partially blocked by selective 5-HT2A (ketanserin; 10 μg/paw) and 5-HT3 (ondansetron; 10 μg/paw) receptor antagonists but not by a selective 5-HT1B receptor antagonist (isamoltan; 10 μg/paw). A non-selective opioid receptor antagonist (naloxone; 50 μg/paw) completely reversed DMT-induced peripheral antinociception, while selective CB1 (AM251; 80 μg/paw) and CB2 (AM630; 100 μg/paw) cannabinoid receptor antagonists partially blocked this effect. Additionally, a non-selective nitric oxide synthase inhibitor (L-NOArg; 24 μg/paw) partially reversed DMT’s antinociceptive effect, which remained unaffected by the selective ATP-sensitive calcium channel blocker (glibenclamide; 80 μg/paw). These findings suggest that DMT exhibits peripheral antinociceptive activity, involving opioid receptors, CB1 and CB2 cannabinoid receptors, 5-HT2A and 5-HT3 serotonergic receptors, and NOS enzymes.
Keywords
Serotonin, Cannabinoid, Opioid, Nitric oxide, Pain
Abbreviations
5-MeO-DMT: 5-methoxy-N,N-dimethyltryptamine; AEA: Anandamide; ANOVA: Analysis of Variance; CaCCs: Calcium-activated Chloride Channels; DMT: N,N-dimethyltryptamine; DMSO: Dimethyl Sulfoxide; GABA A: γ-Aminobutyric Acid A; Glib: Glibenclamide; ISA: Isamoltan; Ket: Ketanserin; NO: Nitric Oxide; NOS: NO Synthase; NX: Naloxone; Onda: Ondansetron; PEA: N-palmitoylethanolamine; PGE2: Prostaglandin E2; S.E.M.: Standard Error of the Mean; SERT: Serotonin Transporter; VMAT: Monoaminergic Vesicle
Introduction
Ayahuasca is an Amazonian herbal infusion made by boiling the Banisteriopsis caapi vine and the leaves of the shrub Psychotria viridis [1]. Research on populations that regularly use ayahuasca for spiritual reasons shows it is fairly safe and may help improve mental well-being by reducing signs of mental health problems and enhancing cognitive performance on tasks like the Stroop and Wisconsin Card Sorting Tests [2]. In addition to its strong antidepressant and anxiety-reducing effects in humans [3], ayahuasca has also been tested in several behavioral settings relevant to depression and anxiety in rodents [4].
Psychotria viridis contains significant amounts of N,N-dimethyltryptamine (DMT). A considerable amount of evidence suggests that DMT is produced by various animals, including humans [5]. Recent studies in rodents have shown that DMT exists in the brain at levels comparable to those of key neurotransmitters such as serotonin and dopamine [6]. There is growing recognition that endogenous DMT may play a natural role in brain neurochemistry that is not yet fully understood but is known to exist. It could function as a neurotransmitter, neurohormone, or neuromodulator, and might also be involved in other physiological processes, including acting as a signaling molecule or hormone outside the brain [7]. In this context, the authors further suggest that DMT is not present in the brain solely to induce or cause hallucinations.
In fact, when administered experimentally to rodents, it was found to reduce apoptosis and decrease inflammation [8]. DMT has also shown neuroprotective effects and promoted the growth of new neurons in the mice's granular zone [9]. Furthermore, the mice performed better on memory tests compared to controls. Additionally, DMT has demonstrated neuroprotective effects in a rat model of forebrain ischemia and in a mouse model of Alzheimer’s disease [10].
Additionally, DMT produces antidepressant and anxiolytic effects in rodents, encouraging further research as a potential treatment for depression and post-traumatic stress disorder [11]. Notably, this study shows that DMT at a subhallucinogenic dose of 1 mg/kg causes only antidepressant and anxiolytic effects, while 10 mg/kg, a higher dose, leads to effects typical of hallucinogens.
Currently, the analgesic effects of psychoactive agents like DMT are being studied in chronic pain conditions [12]. Recently, our group demonstrated the antinociceptive effect of Psychotria viridis, which contains DMT, on carrageenan-induced hyperalgesia in rats [13]. Therefore, there is strong evidence suggesting that DMT has potential analgesic effects. This study aimed to evaluate the effects of DMT in a nociceptive pain animal model.
This study aimed to evaluate the antinociceptive effects of DMT using the paw pressure test in rats. It also investigated the mechanisms of action of this compound, particularly involving serotonin, opioid, cannabinoid, and nitric oxide/potassium channel systems.
Methods
Animals
Experiments were conducted on male Wistar rats weighing 180-200 g, obtained from the Bioterism Center of the Federal University of Minas Gerais (CEBIO-ICB/UFMG). The animals were kept in a temperature-controlled room (24 ± 1°C) on a 12-hour light/dark cycle (07:00 - 19:00) with free access to food and water. After the experiments, the animals were euthanized. All procedures and protocols involving animals were approved by the Ethics Committee on the Use of Animals at the Federal University of Minas Gerais (protocol nº 58/2019) and follow the guidelines for the assessment of experimental pain in animals [14].
Measurement of hyperalgesia
Hyperalgesia was induced by subcutaneously injecting prostaglandin E2 (PGE2; 2 µg/paw) into the plantar surface of the hind paw, and it was measured using the mechanical paw pressure test described by Randall and Selitto [15]. An analgesy-meter (Ugo-Basile, Italy), consisting of a cone-shaped paw presser with a rounded tip that applies linearly increasing pressure to the hind paw, was used. The nociceptive threshold was determined as the weight in grams required to elicit paw withdrawal, indicating a nociceptive response. A cutoff value of 300 g was set to minimize paw damage. The nociceptive threshold was measured in the right hind paw and calculated as the average of three consecutive trials, recorded before (baseline nociceptive threshold) and after PGE2 injection. The results were calculated as the difference between these two averages (Δ of nociceptive threshold) in grams. To reduce stress, the rats were habituated to the apparatus one day before the experiments.
Experimental protocol
In all protocols, the baseline threshold of each animal was first determined before injecting any substance and after 180 minutes of PGE2 (peak of hyperalgesia). First, the temporal development of the dose-response curve of N,N-dimethyltryptamine (DMT) against PGE2-induced hyperalgesia was assessed. DMT was injected into the right hind paw, and the nociceptive threshold was measured in the same paw before injection (at time zero) and 180 minutes after PGE2 injection (Figure 1A). The difference between these values was expressed as the Δ of the nociceptive threshold.
To exclude systemic effects of DMT, PGE2 was injected into both hind paws, while the highest dose of DMT was injected only into the right paw, with the contralateral paw receiving vehicle. In this case, nociceptive thresholds were measured in both hind paws before injection (at time zero) and 180 minutes after PGE2 injection (Figure 1B).
To investigate the mechanisms of the antinociceptive effect of DMT, serotonin receptor antagonists (isamoltan, ketanserin, and ondansetron) were injected 170 minutes and DMT 145 minutes after PGE2 injection (Figure 1C). To test whether naloxone or L-NOArg (Figures 1D and 1F) could inhibit this effect, these substances were administered 150 minutes and DMT 145 minutes after PGE2 injection. The cannabinoid antagonists (AM251 and AM630) were injected 170 minutes and DMT 145 minutes after PGE2 injection (Figure 1E). Glibenclamide was administered 175 minutes and DMT 145 minutes after PGE2 injection (Figure 1F). The protocols for dose and injection times for each drug used in this study were selected from experiments previously conducted by our group [16]. The experiments were conducted in a double-blind manner, where the person who injected the solutions was not the same as the one who performed the nociceptive behavioral assessment.
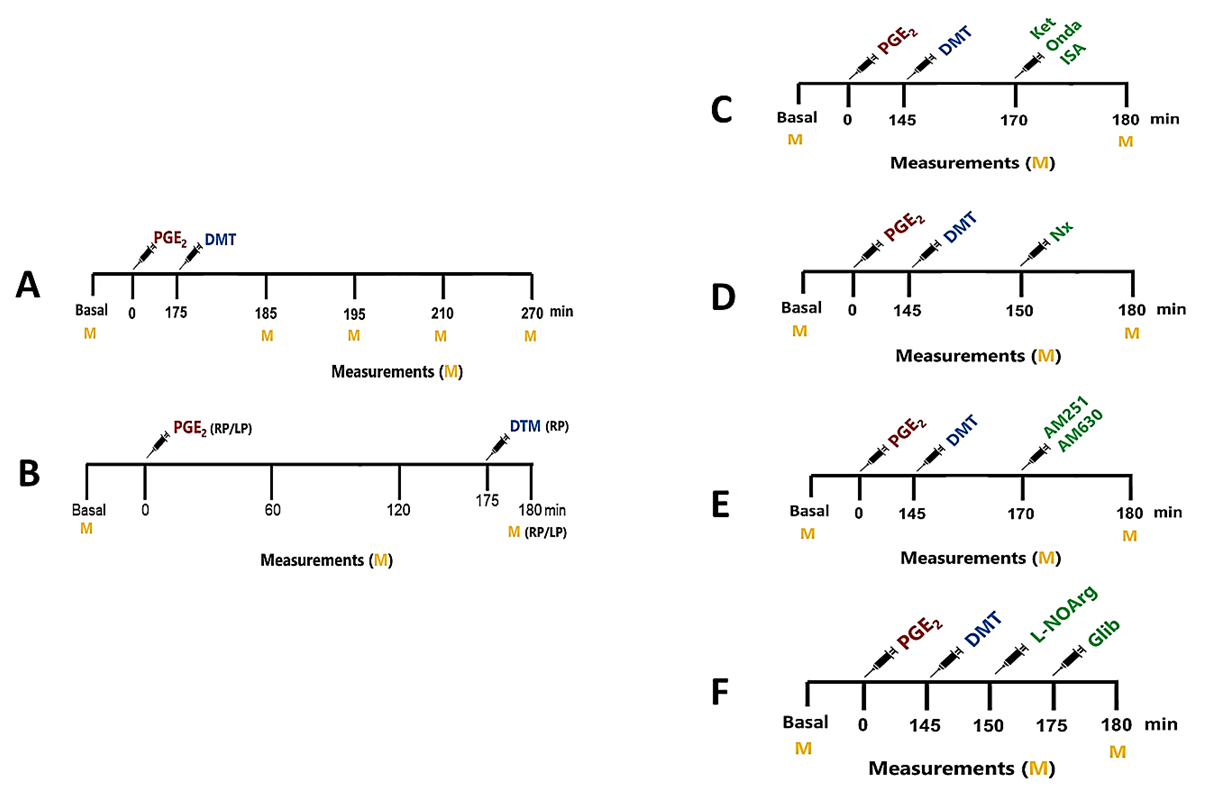
Figure 1. Experimental protocols for evaluating the antinociceptive effects of DMT and its mechanism of action. Assessment of the antinociceptive effect at different time points (A). Exclusion of systemic antinociceptive effects (B). PGE2 was administered at 0 minutes to the right (RP) and left (LP) hind legs of the animals, with DMT at its maximum doses given 175 minutes later only in the RP and with its vehicle in the LP. Nociceptive threshold measurements were taken in both paws before and 180 minutes after PGE2 injection, when the maximum effect occurs. Protocols for using pharmacological tools to evaluate the involvement of the serotonin (C), opioid (D), cannabinoid (E), and nitric oxide (F) systems in DMT-induced antinociception.
Drug administration
DMT was diluted with 10% dimethyl sulfoxide (DMSO), supplied by Federal Police expert André Dias Cavalcanti. Prostaglandin E2 (Sigma, USA), the hyperalgesic agent, was diluted with 10% ethanol. The nonselective opioid antagonist naloxone (Sigma, USA), the nonselective NO synthase inhibitor L-NOArg (RBI, USA), and the selective serotonin antagonists 5-HT1B isamoltan (Tocris, USA), 5-HT2A ketanserin (Biotrend, Switzerland), and 5-HT3 ondansetron (Cristália, Brazil) were diluted in sterile physiological saline. The CB1 cannabinoid antagonist AM251 (Sigma, USA), the CB2 cannabinoid antagonist AM630 (Sigma, USA), and the selective ATP-sensitive K+ channel blocker glibenclamide (Sigma, USA) were dissolved in 10% DMSO. All these drugs were injected into the right plantar surface of the paw at a volume of 50 µL per paw, except for PGE2, which was 100 µL per paw. For injections, the animals were manually restrained.
Statistical analysis
The calculated sample size was 4 (N = 4). The total number of rats (96) was randomly divided into experimental and control groups. Results were expressed as the mean ± standard error of the mean (S.E.M.). Statistical analysis was performed using GraphPad Prism (version 8.0.2). Differences between groups were analyzed with a one-way ANOVA followed by the post hoc Bonferroni test. Data showing a time dependency (Figure 2) were analyzed using a two-way repeated-measures ANOVA, as nociceptive threshold measures for each group were obtained from the same animals over time. Statistical significance was set at p<0.01.
Results
Intraplantar injection of DMT (2.5, 10, and 40 μg/paw) into the right hind paw of the animals elicited a statistically significant antinociceptive response against PGE2 (2 μg/paw)-induced hyperalgesia + DMSO. An increase in the nociceptive threshold was observed 5 minutes after DMT injection (180 minutes after PGE2 injection), with the peak effect occurring 35 minutes after DMT injection, corresponding to 210 minutes after PGE2 administration. Additionally, the 10 μg dose produced intermediate antinociception, whereas the 40 μg dose resulted in maximum antinociception. Conversely, the 2.5 μg dose did not produce a statistically significant difference in nociceptive threshold compared to the control group (PGE2 + vehicle; Figure 2). One-way ANOVA + Bonferroni post hoc test revealed significant main effects of time [F(8,96) = 101.8, P < 0.0001] and drug [F(3,12) = 16.53, P<0.0001].
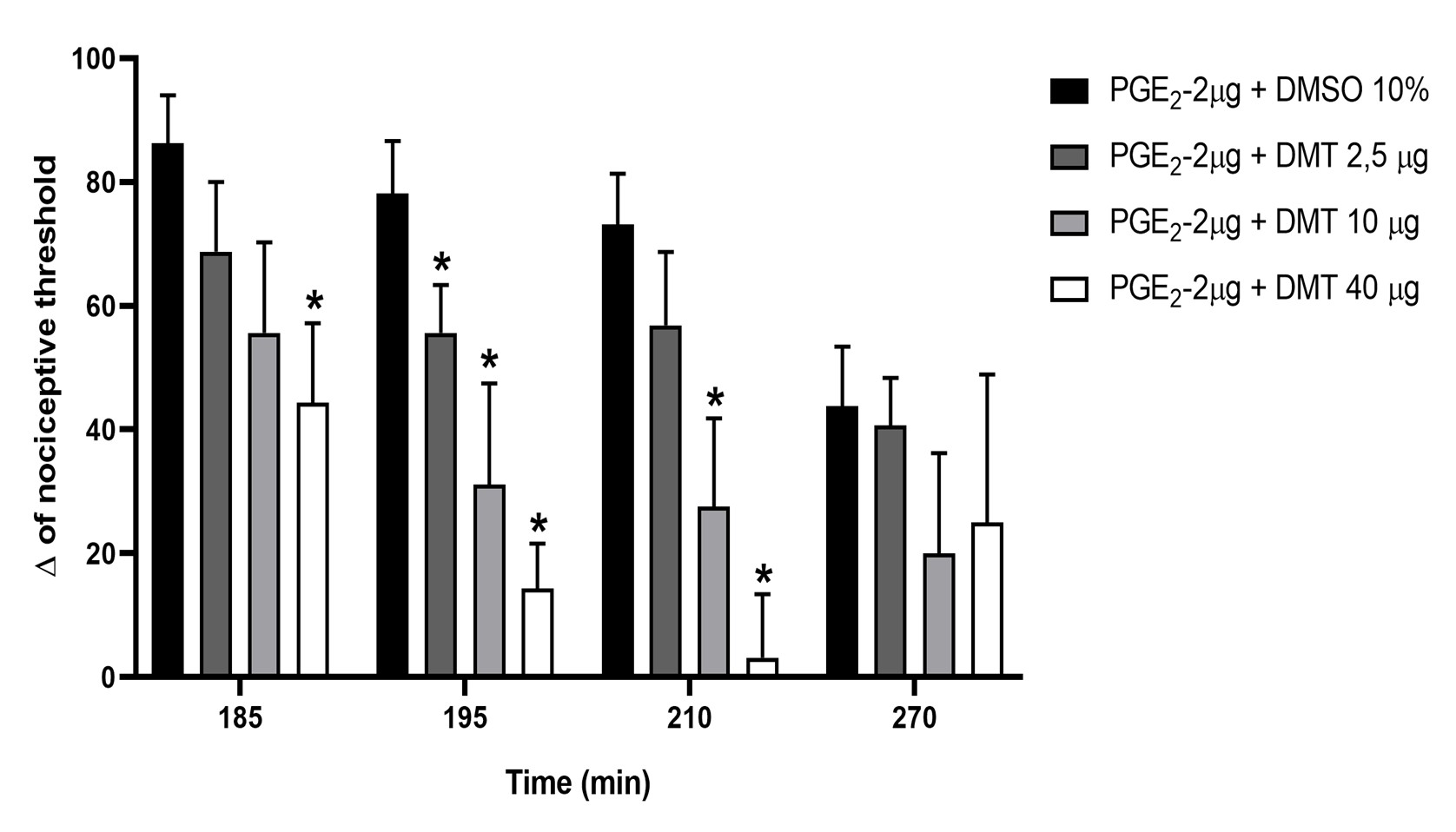
Figure 2. Effect of intraplantar injection of DMT on PGE2-induced hyperalgesia in rats. Prostaglandin E2 (PGE2; 2 μg/paw) and N,N-dimethyltryptamine (DMT; 2.5, 10, and 40 μg/paw) were injected into the right hind paw at 0 and 175 minutes, respectively. Nociceptive threshold measurements were taken at 185, 195, 210, and 270 minutes in the same animals. Each column shows the mean ± S.E.M. (n = 4, total = 16). * indicates a significant difference compared with PGE2 + DMSO 10% (P<0.01), using two-way repeated measures ANOVA followed by the Bonferroni test.
To rule out a potential systemic antinociceptive effect, the highest dose of DMT (40 µg/paw) was injected into the right hind paw (ipsilateral to the nociceptive threshold measurement), and an antinociceptive effect was observed compared to the control group (statistically significant). Conversely, injecting DMT into the contralateral hind paw did not produce antinociception. DMT-induced antinociception was confined to the treated paw, as it did not influence PGE2-induced hyperalgesia in the contralateral paw [F(3,12) = 77.74, P<0.503], indicating that DMT had only a local effect (Figure 3).
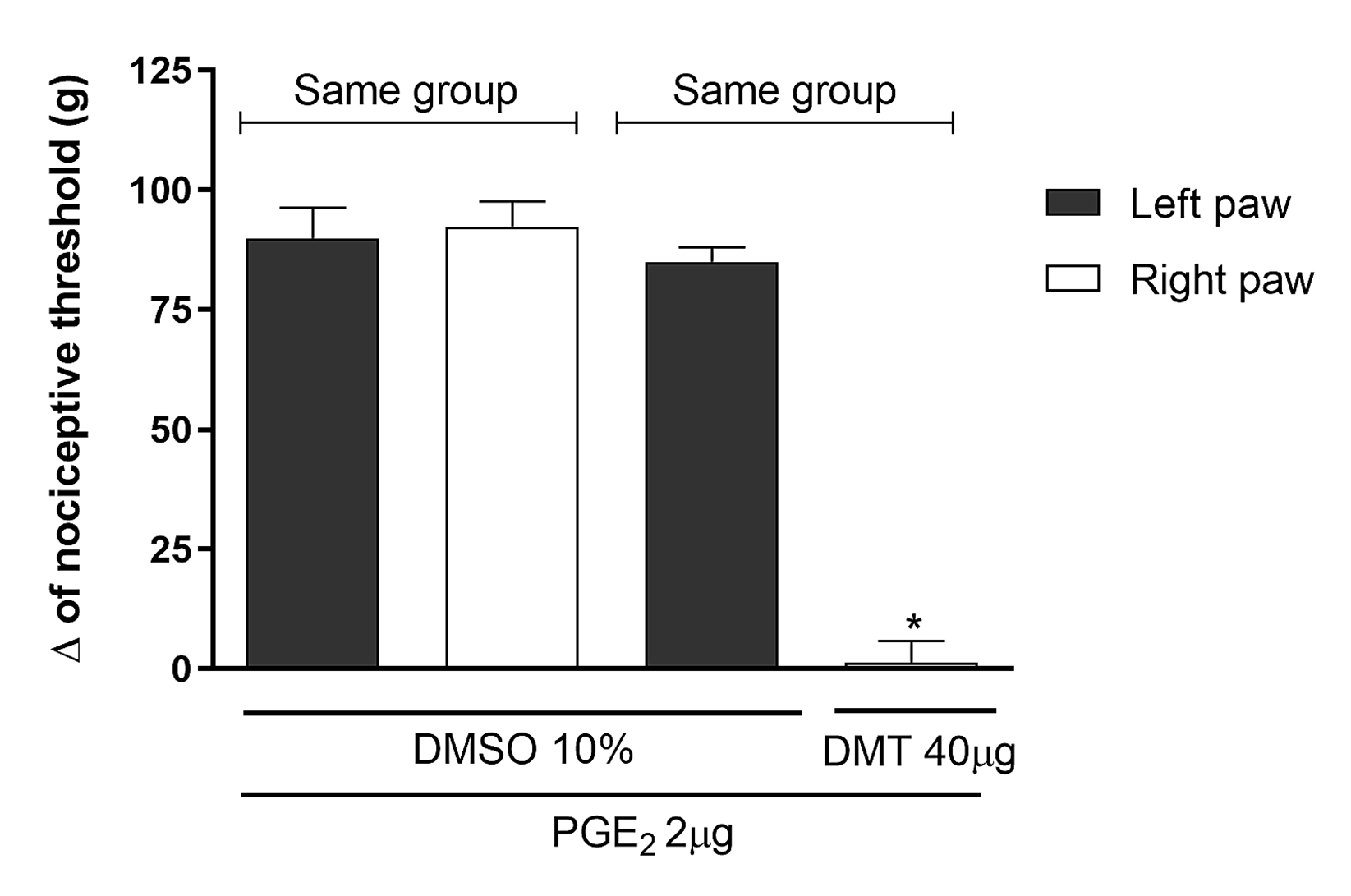
Figure 3. Exclusion of the central or systemic antinociceptive effect of the highest dose of DMT. N,N-dimethyltryptamine (DMT; 40 μg/paw) was administered to the right hind paw 145 minutes after Prostaglandin E2 (PGE2; 2 μg/paw) administration into both hind paws, and nociceptive threshold was measured in either the right (ipsilateral; IL) or the left (contralateral; CL) hind paw. Measurements were taken in both paws at 180 minutes. Each column shows the mean ± S.E.M. (n = 4, total = 8). * indicates a significant difference compared to PGE2 + DMSO 10% (P<0.01), using one-way ANOVA followed by the Bonferroni test.
Since DMT showed significant antinociceptive effects, additional tests were performed to explore the possible mechanism of action of the studied compound. Known antagonists of each pathway involved, whether or not directly related to nociception, were used to determine whether blocking these pathways could reverse DMT's activity. Ketanserin (Ket; 10 μg/paw) and ondansetron (Onda; 10 μg/paw), selective antagonists of 5-HT2A and 5-HT3 receptors, respectively, were injected into the right hind paw of rats. These treatments partially counteracted the peripheral antinociceptive effect of DMT (40 µg/paw) (Figure 4), with significance [F(2,9) = 37.64, p<0.0001] and [F(2,9) = 68.02, p<0.0001]. In contrast, isamoltan (ISA; 10 μg/paw), a selective 5-HT1B receptor antagonist, did not modify [F(2,9) = 79.36, p<0.444] the peripheral antinociceptive effect of DMT at 40 μg/paw (Figure 4).
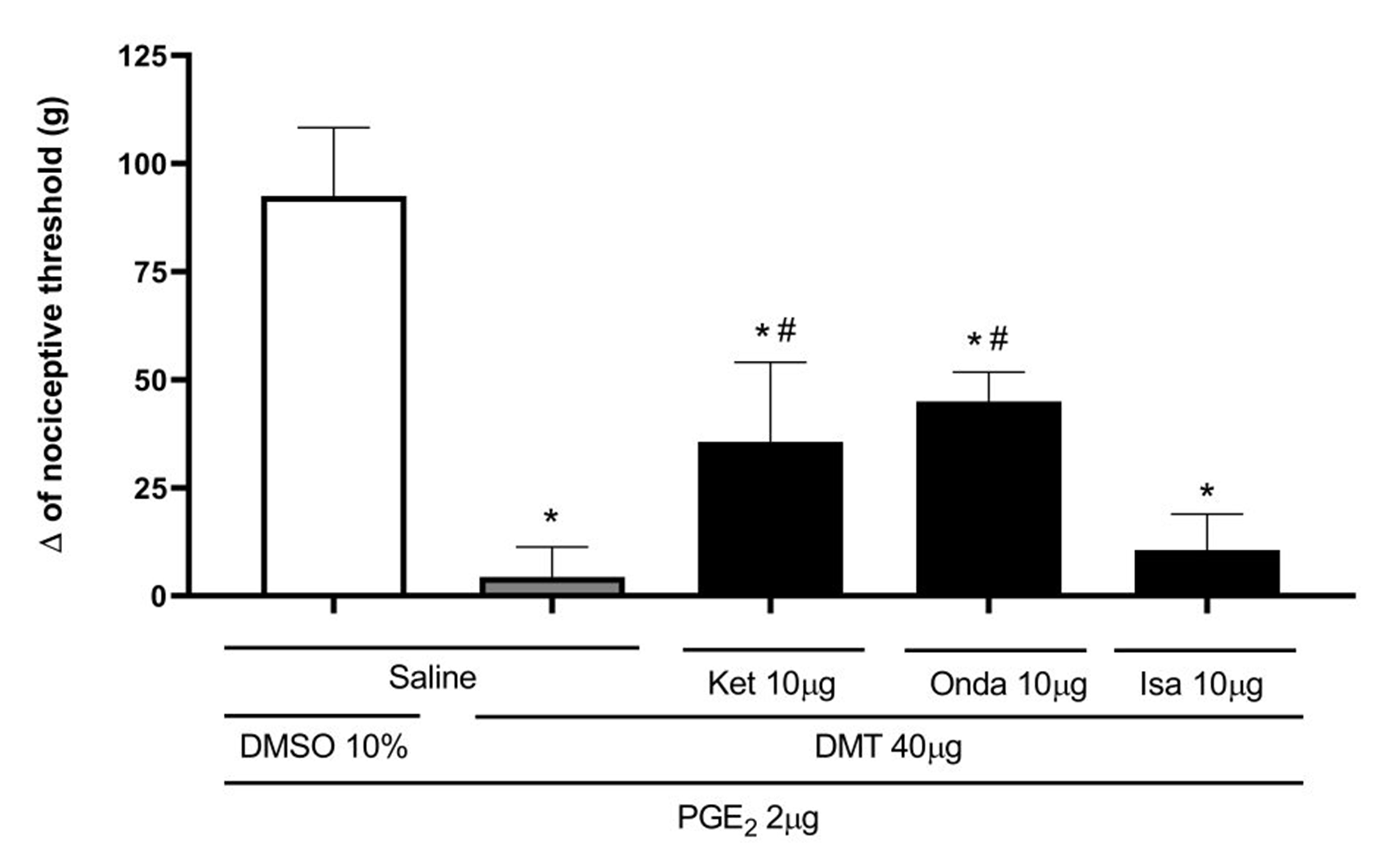
Figure 4. Effect of ketanserin, ondansetron, and isamoltan on DMT-induced antinociceptive effect against hyperalgesia induced by PGE2. Prostaglandin E2 (PGE2; 2 μg/paw), N,N-dimethyltryptamine (DMT; 40 μg/paw), ketanserin (Ket; 10 μg/paw), ondansetron (Onda; 10 μg/paw), and isamoltan (ISA; 10 μg/paw) were injected into the right hind paw at 0, 145, 170, 170, and 170 minutes, respectively. Measurements were taken at 180 minutes. Each column shows the mean ± S.E.M. (n = 4, total = 20). * P<0.05 compared to PGE2 + DMSO 10% + Saline, and # P<0.01 compared to the PGE2 + DMT + Saline; one-way ANOVA followed by the Bonferroni test.
To determine if DMT's effect involves the opioid pathway, the test was performed using naloxone, a non-selective opioid receptor antagonist. Figure 5 shows that naloxone (NX; 50 μg/paw) fully reversed the peripheral antinociceptive effect of DMT [F(2,9) = 80.76, P<0.0001].
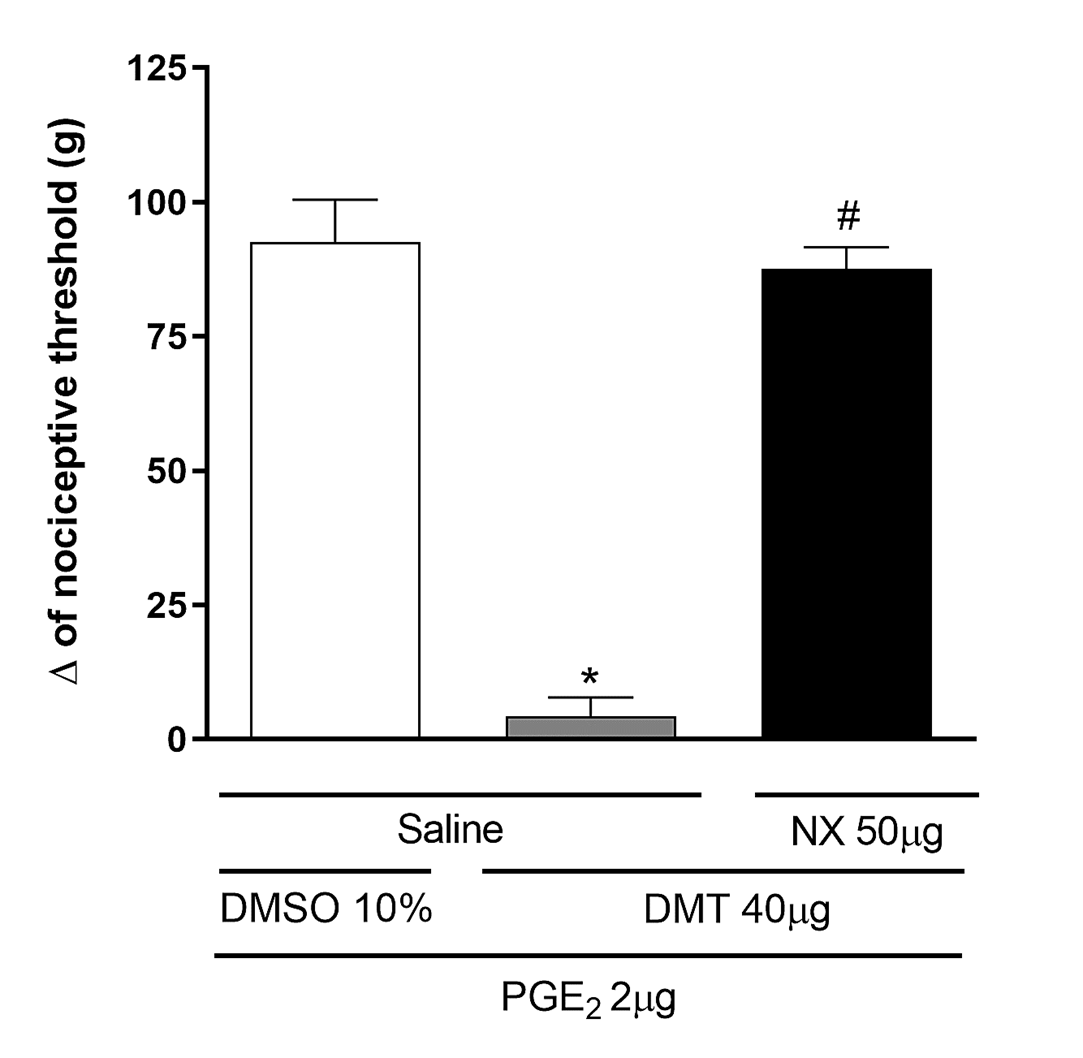
Figure 5. Naloxone antagonizes the DMT-induced antinociceptive effect against PGE2-induced hyperalgesia. Prostaglandin E2 (PGE2; 2 μg/paw), N,N-dimethyltryptamine (DMT; 40 μg/paw), and naloxone (NX; 50 μg/paw) were injected into the right hind paw at 0, 145, and 150 minutes, respectively. Measurements were taken at 180 minutes. Each column shows the mean ± S.E.M. (n = 4, total = 12). * P<0.01 compared to PGE2 + DMSO 10% + Saline and # P<0.05 compared to PGE2 + DMT + Saline; one-way ANOVA followed by the Bonferroni test.
AM251 (80 μg/paw) and AM630 (100 μg/paw), selective CB1 and CB2 cannabinoid receptor antagonists, respectively, partially antagonized DMT-induced peripheral antinociception [F(2,9) = 110.3, P<0.0001] and [F(2, 9) = 92.40, P<0.0001] (Figure 6). Similarly, L-NOArg (24 μg/paw), a non-selective nitric oxide (NO) synthase inhibitor, partially reversed the antinociceptive effect of DMT [F(2,9) = 65.46, P < 0.0001] (Figure 7). In contrast, Figure 7 also shows that intraplantar injection of glibenclamide (80 μg/paw), a selective ATP-sensitive K+ channel blocker, was unable to reverse the antinociceptive effect of DMT [F(2,9) = 80.42, p < 0.2037].
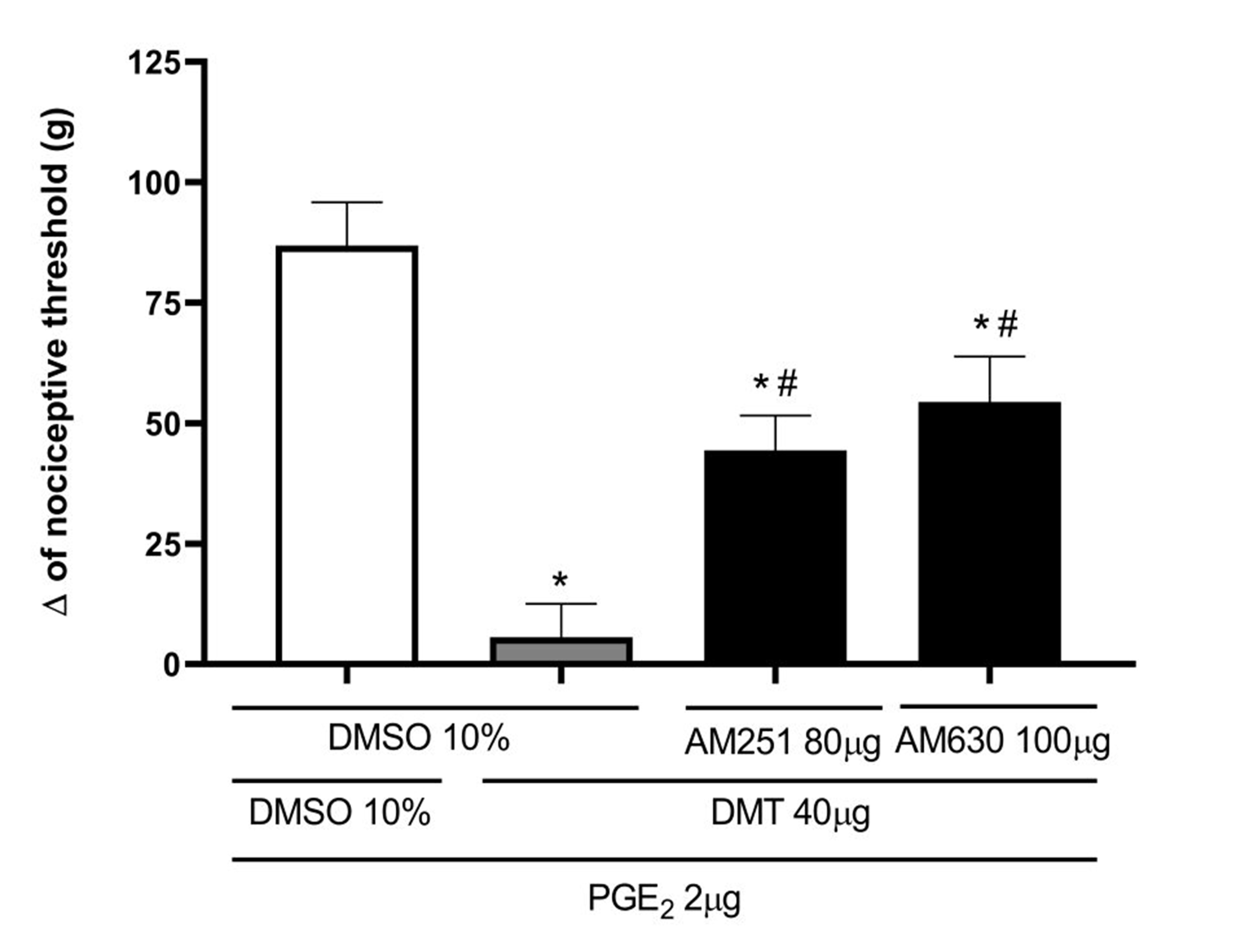
Figure 6. AM251 and AM630 antagonize the DMT-induced antinociceptive effect against PGE2-induced hyperalgesia. Prostaglandin E2 (PGE2; 2 μg/paw), N,N-dimethyltryptamine (DMT; 40 μg/paw), AM251 (80 μg/paw), and AM630 (100 μg/paw) were injected into the right hind paw at 0, 145, 170, and 170 minutes, respectively. Measurements were taken at 180 minutes. Each column shows the mean ± S.E.M. (n = 4, total = 16). * P<0.05 compared to the PGE2 + DMSO 10% + DMSO 10%, and # P<0.01 compared to the PGE2 + DMT + DMSO 10%; one-way ANOVA followed by the Bonferroni test.
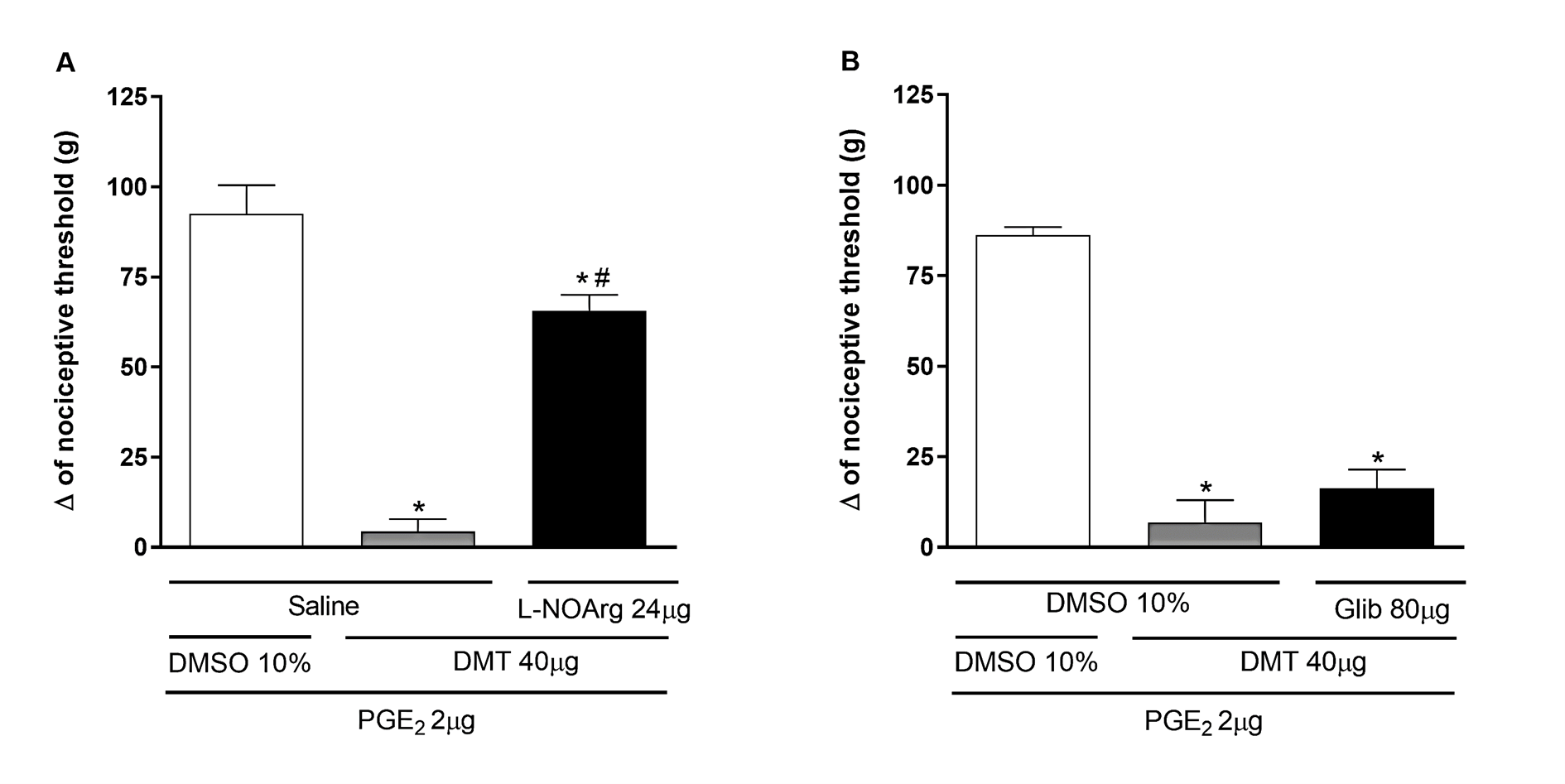
Figure 7. Effect of L-NOArg and glibenclamide on DMT-induced antinociception against PGE2-induced hyperalgesia. Prostaglandin E2 (PGE2; 2 μg/paw), N,N-dimethyltryptamine (DMT; 40 μg/paw), L-NOArg (panel A; 24 μg/paw), and glibenclamide (Glib; panel B; 80 μg/paw) were injected into the right hind paw at 0, 145, 150, and 175 minutes, respectively. Measurements were taken at 180 minutes. Each column shows the mean ± S.E.M. (n=4, total=24). * P<0.01 compared to PGE2 + DMSO 10% + Saline/DMSO 10%, and # P<0.05 compared to PGE2 + DMT + Saline/DMSO 10%; one-way ANOVA followed by the Bonferroni test.
Discussion
In this study, we investigated whether DMT provides peripheral pain relief by reducing the nociceptive response induced by PGE2, a key mediator of pain and inflammation. Prostaglandin is known to cause hyperalgesia by directly sensitizing nociceptors on primary afferent terminals [17]. PGE2-induced hyperalgesia is considered a more reliable model of inflammatory hyperalgesia than other models, such as carrageenan, because it avoids interference from various other mediators produced during inflammation [18]. Our results showed that intraplantar injection of DMT (40 μg/paw) reversed PGE2-induced hyperalgesia. Additionally, the pain-relieving effect of DMT was dose-dependent and limited to the treated paw, indicating a local effect at the doses used in this study rather than a systemic one. To our knowledge, this is the first study to demonstrate the peripheral pain-relief effect of this compound.
Once the peripheral antinociceptive action of DMT was confirmed, its mechanism of action was clarified. Previous studies have shown that DMT acts as a substrate for serotonin transporters (SERT) and monoaminergic vesicle transporters (VMAT) [19–21]. Additionally, it has been demonstrated that DMT binds to serotonergic receptors due to its structural similarity to serotonin [22]. Therefore, we examined the involvement of the serotonergic system in DMT’s peripheral antinociceptive effect using selective 5-HT2A (ketanserin), 5-HT3 (ondansetron), and 5-HT1B (isamoltan) receptor antagonists. Our results indicated that ketanserin and ondansetron partially reversed DMT’s antinociceptive effect, suggesting that 5-HT2A and 5-HT3 receptors are involved in DMT-induced peripheral antinociception. It was demonstrated that 5-HT2A, 5-HT3, and 5-HT1B receptors are involved in serotonin-induced peripheral antinociception, as selective antagonists for each receptor reversed this effect [23]. Supporting our findings, it has been shown that DMT acts as an agonist at 5-HT2A receptors, which are responsible for its hallucinogenic effects [20,24] demonstrated that intrathecal administration of 5-methoxy-N,N-dimethyltryptamine (5-MeO-DMT), a methoxylated derivative of DMT, produces an antinociceptive effect that was blocked by 5-HT3 and GABA-A receptor antagonists but not by antagonists for 5-HT2, 5-HT1A, 5-HT1B, or 5-HT1S receptors. However, DMT also has an affinity for 5-HT1A / 1B / 1D / 2B / 2C / 6 / 7 receptors, while having partial agonist activity at 5-HT1A / 2A / 2C receptors [25–27].
Since the reversal of analgesia by serotonergic receptor antagonism was only partial, other systems might also be involved in DMT's analgesic effect. Therefore, we examined the opioid and cannabinoid systems. A non-selective opioid receptor antagonist (naloxone) was used, which reversed DMT-induced peripheral antinociceptive effects, indicating the involvement of the opioid system. Likewise, opioids have been shown to interfere with DMT. To our knowledge, there is only one study linking the two systems which demonstrated that pretreatment with opioid agonists and antagonists altered the effects of DMT-induced disruption of food-rewarded bar-pressing behavior in rats [28].
There is substantial scientific evidence indicating that several analgesic agents work by triggering the release of endogenous opioids, such as xylazine and ketamine [29,30]. Regarding the last drug, our research team identified keratinocyte activation and subsequent beta-endorphin release from these cells as mechanisms behind peripheral ketamine-induced analgesia. In relation to DMT, further studies are needed to identify which opioid peptide should be released and its source.
For many years, evidence has accumulated suggesting that cannabinoids can produce antinociception through peripheral mechanisms involving CB1 and CB2 receptors [31]. To determine whether the cannabinoid system contributes to the peripheral antinociceptive effects of DMT, we examined the roles of CB1 and CB2 cannabinoid receptors. AM251, AM630, CB1, and CB2 receptor antagonists were used in this study. A previous study demonstrated that these antagonists, in a dose-dependent manner, blocked the peripheral antinociceptive effects of anandamide (AEA), a CB1 agonist, and N-palmitoyl ethanolamine (PEA), a CB2 agonist, confirming their suitability for this purpose [32]. These selective CB1 (AM251) and CB2 (AM630) antagonists partially reduced DMT-induced peripheral antinociceptive effects, indicating the involvement of CB1 and CB2 receptors. Our results align with known receptor locations, as previous research has shown that CB1 receptors are densely expressed in the dorsal horn, dorsal root ganglion, and peripheral terminals of primary afferent neurons [33]. Likewise, CB2 receptors are found in immune cells, dorsal root ganglia, and peripheral neurons [34].
A significant interaction between the endogenous opioid and cannabinoid systems in pain management [35,36] has been documented in the literature. Although opioids and cannabinoids bind to different receptors, one system can enhance the other, suggesting that these systems may work synergistically [35]. Additionally, behavioral and molecular studies have shown that cannabinoids induce the release of endogenous opioids, just as opioids can trigger the release of endocannabinoids [37,38].
Our research group demonstrated that peripheral antinociception induced by various analgesic drugs, including opioids and cannabinoids, ultimately involves activation of the nitric oxide (NO) and potassium channel pathway [39,40]. Therefore, we examined the role of the arginine/NO/cGMP pathway in DMT-induced antinociception against PGE2 using the non-selective NO synthase (NOS) inhibitor L-NOArg. Pretreatment with this inhibitor partly reversed the DMT-induced peripheral antinociception, indicating that NO is involved. Activation of the arginine/NO/cGMP pathway is known to produce both central [41] and peripheral [42] analgesia, and has previously been linked to the activation of potassium (K+) channels in its peripheral effects [43,44].
Therefore, we examined the effects of glibenclamide, a selective blocker of ATP-sensitive K+ channels (KATP). Our results showed that this compound did not affect the antinociceptive effect of DMT, indicating that KATP channels do not mediate this effect. Conversely, the peripheral antinociceptive effect of serotonin occurs through activation of the cannabinoid system, which subsequently activates the NO/cGMP/KATP pathway [45]. However, consistent with our results, it was shown that K+ channels are not involved in the peripheral antinociception of bremazocine, despite this drug activating the NO/cGMP pathway [46]. Additionally, it has been demonstrated that activation of opioid [47,48] and cannabinoid [49] receptors induces antinociception through calcium-activated chloride channels (CaCCs). Based on these findings, we suggest that the antinociceptive effect of DMT, via activation of the arginine/NO/cGMP pathway without involving KATP channels, might involve CaCCs, considering the role of opioids and cannabinoids in this process. Future research will be needed to explore this hypothesis.
A key point to discuss is that DMT has a hallucinogenic effect [50]. Another substance with hallucinogenic effects is ketamine, which has been widely used as an anesthetic and analgesic [51], providing an important example that opens the perspective for investigating the therapeutic potential of DMT. It is vital to highlight the existence of several strategies to reduce the Central Nervous System (CNS) effects, such as: a) exploring whether hallucinogenic and analgesic effects occur at different dose ranges. Evidence from rodent drug discrimination studies suggests that DMT at a dose of 10 mg/kg produces effects typical of hallucinogens, while 1 mg/kg only causes antidepressant and anxiolytic behaviors [11]; b) administering drugs through routes that avoid central supramedullary effects, such as local ketamine for postoperative pain relief after third molar surgery [52]. In this context, our work emphasizes the potential for detecting the peripheral antinociceptive action of DMT; c) pharmaceutical chemistry studies focused on modifying chemical structures to alter their pharmacokinetic distribution to the central nervous system and their pharmacodynamic profiles. This approach was used to develop second-generation antihistamines to limit CNS access [53].
Based on the considerations above, the authors suggest conducting future studies with research groups in various fields, such as chemistry, to develop strategies that utilize the analgesic potential of DMT identified in this work.
Conclusion
In conclusion, this study demonstrated that DMT has peripheral pain-relieving effects and identified part of the mechanism underlying this action. This process involves activating multiple systems, including opioid receptors, CB1 and CB2 cannabinoid receptors, 5-HT2A and 5-HT3 serotonergic receptors, and nitric oxide (NO). It is also concluded that ATP-sensitive potassium channels may not be involved in DMT's antinociceptive effect because a potassium channel blocker, glibenclamide, did not influence it. Further research is needed to explore DMT's potential as an analgesic.
Author Contributions
A.C.P., I.D.G.D., and T.R.L.R. conceived and designed experiments. D.P.D.M and F.C.S.F performed the experiments. I.D.G.D., D.P.D.M., and F.C.S.F. analyzed the data and performed statistical analysis. A.D.C. provided DMT. D.P.D.M. and F.C.S.F. prepared the manuscript. I.D.G.D. provided the overall direction of the project and revised the manuscript accordingly. All authors reviewed, discussed, and approved the manuscript.
Declaration of Interest
All authors declared there is no financial interest.
Funding Statement
This work was supported financially by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), and Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG).
Acknowledgements
The authors would like to thank the funding agencies.
References
2. Bouso JC, González D, Fondevila S, Cutchet M, Fernández X, Ribeiro Barbosa PC, et al. Personality, psychopathology, life attitudes and neuropsychological performance among ritual users of Ayahuasca: a longitudinal study. PLoS One. 2012;7(8):e42421.
3. Sarris J, Perkins D, Cribb L, Schubert V, Opaleye E, Bouso JC, et al. Ayahuasca use and reported effects on depression and anxiety symptoms: An international cross-sectional study of 11,912 consumers. Journal of Affective Disorders Reports. 2021 Apr 1;4:100098.
4. Dos Santos RG, Osório FL, Crippa JA, Hallak JE. Antidepressive and anxiolytic effects of ayahuasca: a systematic literature review of animal and human studies. Braz J Psychiatry. 2016 Mar;38(1):65–72.
5. Barker SA, McIlhenny EH, Strassman R. A critical review of reports of endogenous psychedelic N, N-dimethyltryptamines in humans: 1955-2010. Drug Test Anal. 2012 Jul-Aug;4(7-8):617–35.
6. Jiménez JH, Bouso JC. Significance of mammalian N, N-dimethyltryptamine (DMT): A 60-year-old debate. J Psychopharmacol. 2022 Aug;36(8):905–19.
7. Schimmelpfennig J, Jankowiak-Siuda K. Unique biological and physiological properties of endogenous N, N-dimethyltryptamine from the perspective of functioning of the nervous system. Neuropsychiatria i Neuropsychologia/Neuropsychiatry and Neuropsychology. 2023;18(1):1–10.
8. Nardai S, László M, Szabó A, Alpár A, Hanics J, Zahola P, et al. N,N-dimethyltryptamine reduces infarct size and improves functional recovery following transient focal brain ischemia in rats. Exp Neurol. 2020 May;327:113245.
9. Morales-Garcia JA, Calleja-Conde J, Lopez-Moreno JA, Alonso-Gil S, Sanz-SanCristobal M, Riba J, et al. N,N-dimethyltryptamine compound found in the hallucinogenic tea ayahuasca, regulates adult neurogenesis in vitro and in vivo. Transl Psychiatry. 2020 Sep 28;10(1):331.
10. Barker SA. N, N-dimethyltryptamine (DMT) in rodent brain: Concentrations, distribution, and recent pharmacological data. Prog Neuropsychopharmacol Biol Psychiatry. 2025 Mar 20;137:111259.
11. Cameron LP, Benson CJ, Dunlap LE, Olson DE. Effects of N, N-Dimethyltryptamine on Rat Behaviors Relevant to Anxiety and Depression. ACS Chem Neurosci. 2018 Jul 18;9(7):1582–90.
12. Kooijman NI, Willegers T, Reuser A, Mulleners WM, Kramers C, Vissers KCP, et al. Are psychedelics the answer to chronic pain: A review of current literature. Pain Pract. 2023 Apr;23(4):447–58.
13. Machado PD, Fonseca FCS, Azevedo MC, Filho SAV, Cavalcanti AD, Santos ODH, et al. Antinociceptive effects of Psychotria viridis Ruiz & Pav. on carrageenan-induced hyperalgesia in rats. Curr Top Pharmacol. 27: 1–8.
14. Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983 Jun;16(2):109–10.
15. RANDALL LO, SELITTO JJ. A method for measurement of analgesic activity on inflamed tissue. Arch Int Pharmacodyn Ther. 1957 Sep 1;111(4):409–19.
16. Soares-Santos RR, Machado DP, Romero TL, Duarte IDG. Nitric oxide and potassium channels but not opioid and cannabinoid receptors mediate tramadol-induced peripheral antinociception in rat model of paw pressure withdrawal. Can J Physiol Pharmacol. 2024 Mar 1;102(3):218–27.
17. Wall PD, Melzack R, McMahon SB, Koltzenburg M, Tracey I, Turk DC. Wall and Melzack's textbook of pain. (No Title). 2006.
18. Hata AN, Breyer RM. Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther. 2004 Aug;103(2):147–66.
19. Cozzi NV, Gopalakrishnan A, Anderson LL, Feih JT, Shulgin AT, Daley PF, et al. Dimethyltryptamine and other hallucinogenic tryptamines exhibit substrate behavior at the serotonin uptake transporter and the vesicle monoamine transporter. J Neural Transm (Vienna). 2009 Dec;116(12):1591–9.
20. Halberstadt AL. Recent advances in the neuropsychopharmacology of serotonergic hallucinogens. Behav Brain Res. 2015 Jan 15;277:99–120.
21. Nagai F, Nonaka R, Satoh Hisashi Kamimura K. The effects of non-medically used psychoactive drugs on monoamine neurotransmission in rat brain. Eur J Pharmacol. 2007 Mar 22;559(2-3):132–7.
22. Halman A, Kong G, Sarris J, Perkins D. Drug-drug interactions involving classic psychedelics: A systematic review. J Psychopharmacol. 2024 Jan;38(1):3–18.
23. Diniz DA, Petrocchi JA, Navarro LC, Souza TC, Castor MG, Perez AC, et al. Serotonin induces peripheral mechanical antihyperalgesic effects in mice. Eur J Pharmacol. 2015 Nov 15;767:94–7.
24. Carbonaro TM, Gatch MB. Neuropharmacology of N,N-dimethyltryptamine. Brain Res Bull. 2016 Sep;126(Pt 1):74–88.
25. Fontanilla D, Johannessen M, Hajipour AR, Cozzi NV, Jackson MB, Ruoho AE. The hallucinogen N,N-dimethyltryptamine (DMT) is an endogenous sigma-1 receptor regulator. Science. 2009 Feb 13;323(5916):934–7.
26. Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, et al. Predicting new molecular targets for known drugs. Nature. 2009 Nov 12;462(7270):175–81.
27. Riba J, McIlhenny EH, Valle M, Bouso JC, Barker SA. Metabolism and disposition of N,N-dimethyltryptamine and harmala alkaloids after oral administration of ayahuasca. Drug Test Anal. 2012 Jul-Aug;4(7-8):610–6.
28. Ruffing DM, Domino EF. Effects of selected opioid agonists and antagonists on DMT- and LSD-25-induced disruption of food-rewarded bar pressing behavior in the rat. Psychopharmacology (Berl). 1981;75(3):226–30.
29. Romero TR, Pacheco Dda F, Duarte ID. Xylazine induced central antinociception mediated by endogenous opioids and μ-opioid receptor, but not δ-or κ-opioid receptors. Brain Res. 2013 Apr 19;1506:58–63.
30. Petrocchi JA, de Almeida DL, Paiva-Lima P, Queiroz-Junior C, Caliari MV, Duarte IDG, et al. Peripheral antinociception induced by ketamine is mediated by the endogenous opioid system. Eur J Pharmacol. 2019 Dec 15;865:172808.
31. Vučković S, Srebro D, Vujović KS, Vučetić Č, Prostran M. Cannabinoids and Pain: New Insights From Old Molecules. Front Pharmacol. 2018 Nov 13;9:1259.
32. Romero TR, Resende LC, Guzzo LS, Duarte ID. CB1 and CB2 cannabinoid receptor agonists induce peripheral antinociception by activation of the endogenous noradrenergic system. Anesth Analg. 2013 Feb;116(2):463–72.
33. Veress G, Meszar Z, Muszil D, Avelino A, Matesz K, Mackie K, et al. Characterisation of cannabinoid 1 receptor expression in the perikarya, and peripheral and spinal processes of primary sensory neurons. Brain Struct Funct. 2013 May;218(3):733–50.
34. Anand U, Otto WR, Sanchez-Herrera D, Facer P, Yiangou Y, Korchev Y, et al. Cannabinoid receptor CB2 localisation and agonist-mediated inhibition of capsaicin responses in human sensory neurons. Pain. 2008 Sep 15;138(3):667–80.
35. Cichewicz DL. Synergistic interactions between cannabinoid and opioid analgesics. Life Sci. 2004 Jan 30;74(11):1317–24.
36. Anand P, Whiteside G, Fowler CJ, Hohmann AG. Targeting CB2 receptors and the endocannabinoid system for the treatment of pain. Brain Res Rev. 2009 Apr;60(1):255–66.
37. Ibrahim MM, Porreca F, Lai J, Albrecht PJ, Rice FL, Khodorova A, et al. CB2 cannabinoid receptor activation produces antinociception by stimulating peripheral release of endogenous opioids. Proc Natl Acad Sci U S A. 2005 Feb 22;102(8):3093–8.
38. Welch SP. Interaction of the cannabinoid and opioid systems in the modulation of nociception. Int Rev Psychiatry. 2009 Apr;21(2):143–51.
39. Romero TR, Resende LC, Duarte ID. The neuronal NO synthase participation in the peripheral antinociception mechanism induced by several analgesic drugs. Nitric Oxide. 2011 Nov 30;25(4):431–5.
40. Reis GM, Ramos MA, Pacheco Dda F, Klein A, Perez AC, Duarte ID. Endogenous cannabinoid receptor agonist anandamide induces peripheral antinociception by activation of ATP-sensitive K+ channels. Life Sci. 2011 Apr 11;88(15-16):653–7.
41. Duarte ID, Ferreira SH. The molecular mechanism of central analgesia induced by morphine or carbachol and the L-arginine-nitric oxide-cGMP pathway. Eur J Pharmacol. 1992 Oct 6;221(1):171–4.
42. Durate ID, Lorenzetti BB, Ferreira SH. Peripheral analgesia and activation of the nitric oxide-cyclic GMP pathway. Eur J Pharmacol. 1990 Sep 21;186(2-3):289–93.
43. Rodrigues AR, Duarte ID. The peripheral antinociceptive effect induced by morphine is associated with ATP-sensitive K(+) channels. Br J Pharmacol. 2000 Jan;129(1):110–4.
44. Soares AC, Duarte ID. Dibutyryl-cyclic GMP induces peripheral antinociception via activation of ATP-sensitive K(+) channels in the rat PGE2-induced hyperalgesic paw. Br J Pharmacol. 2001 Sep;134(1):127–31.
45. Aguiar DD, Petrocchi JA, da Silva GC, Lemos VS, Castor MGME, Perez AC, et al. Participation of the cannabinoid system and the NO/cGMP/KATP pathway in serotonin-induced peripheral antinociception. Neurosci Lett. 2024 Jan 1;818:137536.
46. Amarante LH, Alves DP, Duarte ID. Study of the involvement of K+ channels in the peripheral antinociception of the kappa-opioid receptor agonist bremazocine. Eur J Pharmacol. 2004 Jun 28;494(2-3):155–60.
47. Pacheco Dda F, Pacheco CM, Duarte ID. Peripheral antinociception induced by δ-opioid receptors activation, but not μ- or κ-, is mediated by Ca²⁺-activated Cl⁻ channels. Eur J Pharmacol. 2012 Jan 15;674(2-3):255–9.
48. Pacheco Dda F, Pacheco CM, Duarte ID. δ-Opioid receptor agonist SNC80 induces central antinociception mediated by Ca2+ -activated Cl- channels. J Pharm Pharmacol. 2012 Aug;64(8):1084–9.
49. Romero TR, Pacheco Dda F, Duarte ID. Probable involvement of Ca(2+)-activated Cl(-) channels (CaCCs) in the activation of CB1 cannabinoid receptors. Life Sci. 2013 May 2;92(14-16):815–20.
50. Winstock AR, Kaar S, Borschmann R. Dimethyltryptamine (DMT): prevalence, user characteristics and abuse liability in a large global sample. J Psychopharmacol. 2014 Jan;28(1):49–54.
51. Savić Vujović K, Jotić A, Medić B, Srebro D, Vujović A, Žujović J, et al. Ketamine, an Old-New Drug: Uses and Abuses. Pharmaceuticals (Basel). 2023 Dec 21;17(1):16.
52. Esparza-Villalpando V, Ascencio-Padilla R, Pozos-Guillen A, Pozos-Guillen F, Hidalgo-Hurtado JA, Chavarria-Bolaños D. Local Ketamine Improves Postoperative Analgesia After Third Molar Surgery. J Oral Maxillofac Surg. 2019 Dec;77(12):2386–400.
53. Zhang MQ, Ter Laak AM, Timmerman H. Structure-activity relationships within a series of analogues of the histamine H1-antagonist terfenadine. European Journal of Medicinal Chemistry. 1993 Jan 1;28(2):165–73.