Abstract
The formation of new blood vessels, or angiogenesis, is a hallmark of cancer and one of the most important conditions for tumor growth. Vascular endothelial growth factor (VEGF) is one of the most important factors in angiogenesis. Increased VEGF expression has been associated to rapid cancer progression and poor prognosis. The significance of VEGF is not limited to angiogenesis; it also contributes to the development of a tolerogenic tumor microenvironment through a variety of methods. Increased VEGF expression in the tumor microenvironment leads effector immune cells to die while suppressive immune cells become more active. As a result, VEGF has long been recognized as a potent marker of invasive cancer, and inhibiting VEGF signaling can prevent tumor progression and development. Researchers are focusing on medicines and pharmacological agents to prevent neoangiogenesis in different cancers for a long time, mostly by inhibiting VEGF/VEGF-receptor signaling. Anti-VEGF drugs are currently on the market and have been approved for the treatment of various malignancies. They can be used alone or in combination with other targeted therapies like chemotherapy. However, VEGF-signaling inhibition commonly results in the partial clearance of cancer cells, and in certain patients, cancer recurrence may occur, compromising overall survival. In this review, we discussed on VEGF-mediated invasive cancer development, the effect of VEGF on tumor immunity, and therapeutic approaches to combat neoangiogenesis-induced cancer consequences.
Keywords
Cancer, Neoangiogenesis, Immunosuppression, Treg, Tumor microenvironment, VEGF
Introduction
Cancer is one of the most common deadly diseases in the world. Neoplastic transformation of cells requires constant acquisition of mutations in the genome and subsequent transfer of these mutations to the progeny cells which forms a heterogenous population of cancer cells. Hanahan and Weinberg proposed the basic requirements of tumor cells which is required for the successful malignant transformation; popularly known as the hallmarks of cancer. Neoangiogenesis or formation of new blood vessels is one of the crucial hallmarks of cancer progression. Due to unlimited proliferation new cancer cells move further from nearby capillaries limiting the nutrients, growth factors and oxygen supply. Hence, in order to grow beyond a certain size, emerging tumor mass must develop angiogenic ability for limitless supply of nutrients. The formation of new blood vessels necessitates a fine balance of pro- and anti-angiogenic factors. One of the most important growth factors involved in this is vascular endothelial growth factors (VEGFs), more commonly known as VEGFA. Other important angiogenic factors include VEGF isoforms (VEGFB, VEGFC, and VEGFD), platelet-derived growth factor (PDGF), and placenta growth factor [2]. VEGF is a homodimeric glycoprotein with a molecular weight of 45 kDa that binds to the VEGFR (VEGF-receptor) on cellular surfaces [3,4]. Fibroblast growth factor (FGF), epidermal growth factor (EGF), tumor necrosis factor (TNF), transforming growth factor (TGF), and interleukin-1 (IL1) are all known to promote VEGF expression [5,6].
Another potent inducer of VEGF expression is hypoxia. VEGF mRNA expression has been observed to increase in a number of cells in response to decreased oxygen availability. HIF1 (hypoxia-induced factor-1) stabilizes in an oxygen-depleted environment [7]. The factor is ubiquitinated as it undergoes modifications under the action of prolyl-hydroxylase when there is enough of oxygen present. Prolyl-hydroxylase, an oxygen sensor, becomes inactive in the absence of oxygen and so cannot trigger HIF1 ubiquitination [8]. As a result, HIF1 becomes stable in hypoxic conditions and stimulates the production of VEGF and VEGFR [9,10]. Not only hypoxia, but also acidosis, which occurs as a result of inadequate perfusion, causes VEGF expression [3]. HIF-dependent signalling can boost cancer and stromal cell adaptability and selection to their surroundings, leading to changes that favour cancer growth. These circumstances, which are common in the tumor microenvironment, cause the expression of VEGF, which promotes tumor growth.
VEGF binds to two homologous receptors, VEGF receptor-1 and VEGF receptor-2, to exhibit its function [11]. Upon interaction of their ligand, VEGF receptors, like other tyrosine kinase receptors, dimerize, which propagates signal transduction by phosphorylation of the receptors and subsequent signal transduction by particular downstream molecules [12]. A third receptor, VEGF receptor-3, is also present and plays an important role in VEGFC and VEGFD-mediated lymphangiogenesis.
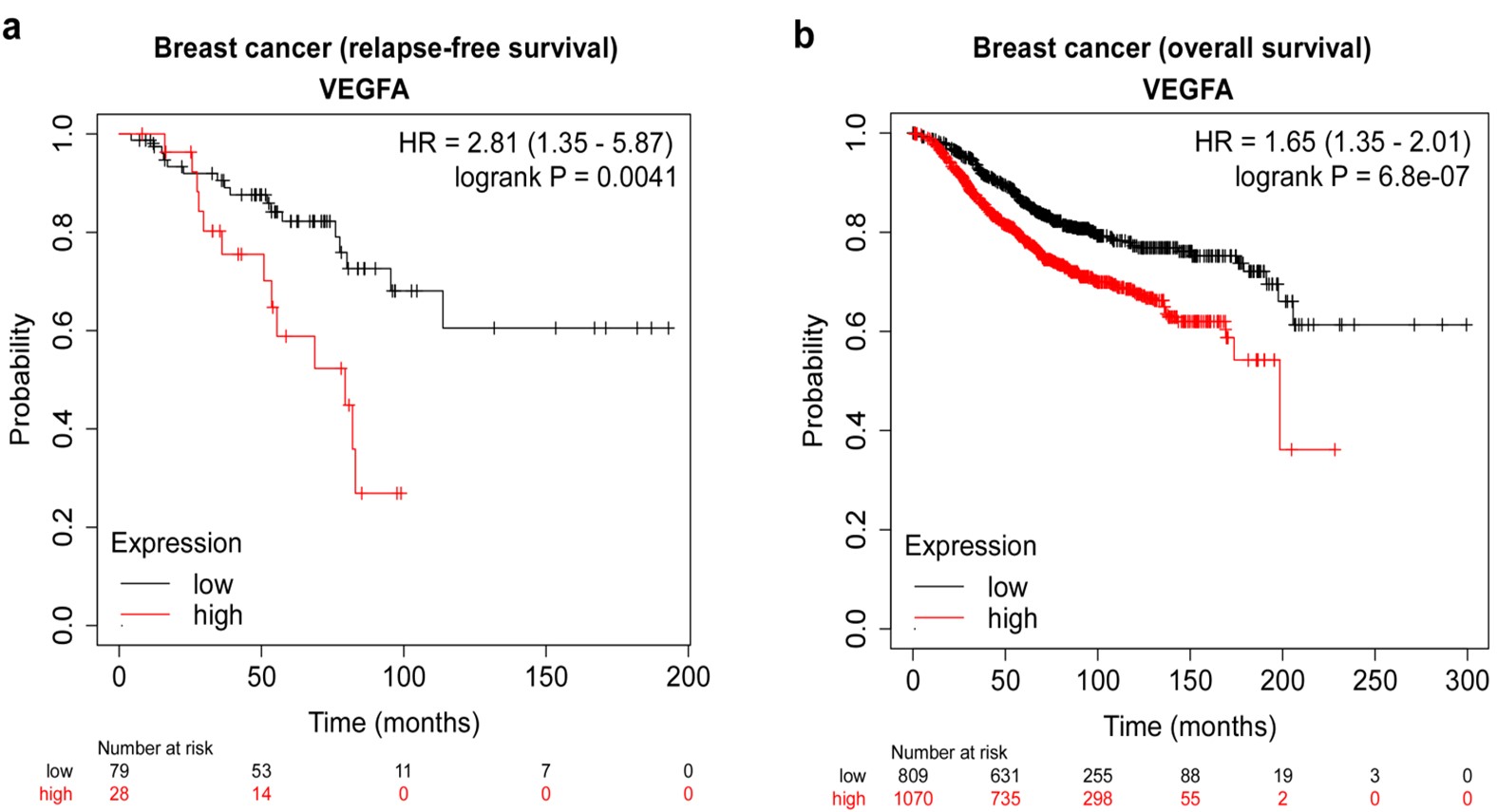
Figure-1: High VEGFA expression is significantly associated with lower survival in breast cancer patients. (a) Kaplan-Meier analysis representing statistical comparison of relapse-free survival between groups with high and low vascular endothelial growth factor-A (VEGFA) mRNA expression levels among patients with hormone receptor-positive (progesterone receptor-positive, human epidermal growth factor receptor-2 (HER2)-positive, breast cancer. (b) Kaplan-Meier analysis representing statistical comparison of overall survival between groups with high and low vascular endothelial growth factor-A (VEGFA) mRNA expression levels among patients in breast cancer.
Sources of VEGF
VEGF, also known as vascular permeability factor (VPF), was originally described as an endothelial cell-specific mitogen. VEGF is produced by many cell types (Figure 2) including:
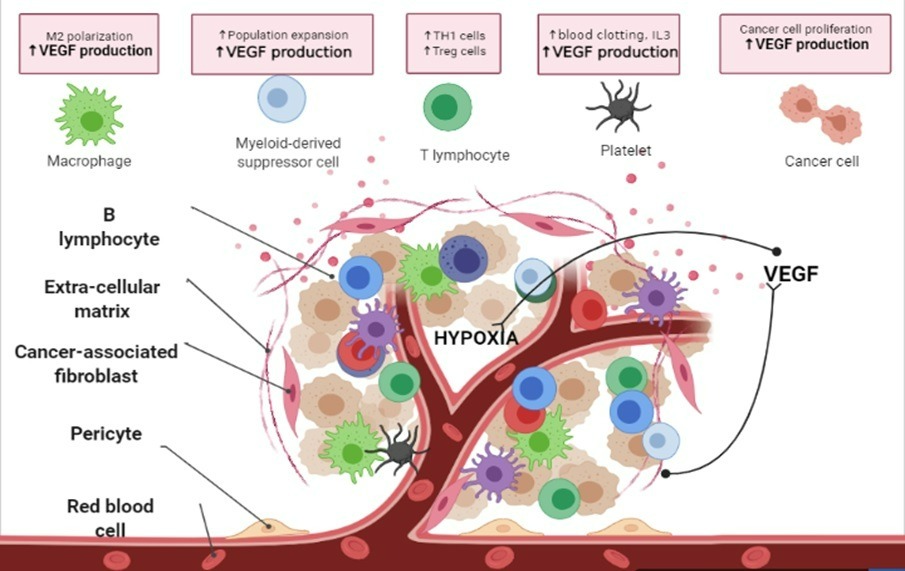
Figure-2: Sources of VEGF: The cellular sources of VEGF are multiple, varying from tumor cells to immune cells like Tregs, macrophages. Fibroblasts, PMN cells, and monocytes (MOs) are sources of VEGF in the wound-healing process. Multiple factors, including hypoxia, nitric oxide, and inflammatory cytokines such as interleukin 3 are involved in controlling VEGF expression in normal tissue repair and remodeling events. (Created in Biorender.com)
Tumor cells
Tumor cells produce VEGF and activate VEGF receptors (VEGFR) on the cell surface, indicating that an autocrine signaling loop exists. VEGF/VEGFR interaction enables cancer cells to promote their growth, survival, and migration by phosphorylation and activation of VEGFR-1/2 or VEGF-induced neuropilin signaling. Because VEGF expression is strongly connected with cancer progression [13], it can be employed as a useful prognostic marker. The plasma concentration of VEGF in cancer patients rises as the disease progresses, indicating that the tumor tissue is the primary source of VEGF [3]. The VEGF gene was genetically ablated in experimental tumor models, and it was observed that VEGF expression and angiogenesis decreased, indicating that tumor cells are the primary source of VEGF. Several studies have highlighted that the main stimulator of VEGF production in tumors is the hypoxic-environment, which is caused by a lack of oxygen due to excessive tumor growth. In various cancers, hypoxia-induced HIF1 regulates VEGF expression. In vivo tumor models with wild-type VEGF genes, however, have revealed that tumor growth is reduced when host fibroblasts do not synthesize VEGF. These findings suggested that VEGF produced by tumor cells is a key factor in neoangiogenesis, but not enough to maintain tumors growing. Recent research has revealed that tumor-associated stromal cells are also key VEGF generators [14,15].
Macrophages
When macrophages adapt to their surroundings and respond with a coordinated set of signals to promote or resolve inflammation, they produce VEGF, which promotes angiogenesis. Various studies have shown that the expression of the VEGF gene in macrophages is regulated by oxygen tension [16]. Though it is not clear whether the expression is targeted at the mRNA level or the stability of the mRNA is targeted. When compared to pro-inflammatory type-I (M1) macrophages, anti-inflammatory tumor-associated type-II macrophages (TAMs/M2) are potent stimulator of tumor growth and secrete VEGF in the TME [17]. By secreting pro-angiogenic and pro-lymphangiogenic molecules (VEGFA, VEGFC, VEGFD, bFGF, TNFa, TGFb, IL1b, PDGFBB, and ECM remodeling molecules including MMPs), pro-angiogenic Tie2+/CD31+ TAMs play a key role in both angiogenesis and lymph-angiogenesis. VEGFA and VEGFC, which are secreted, stimulate mitogenic and chemotactic activities in macrophages, hence increasing TAM activity for effective tumor cell proliferation [18,19]. Multiple tumor types have been linked to increased micro-vessel density in patients with a high-number of TAMs [20]. A direct relationship exists between macrophages and tumor angiogenesis, which is caused by TAM-mediated TME regulation.
Treg cells
Treg cells, characterized by CD4, CD25, FOXP3 signature molecules, secret a cascade of cytokines that are indispensable for their immunosuppressive properties [21,22]. Along with these cytokines, these cells also secrete VEGFA, which is necessary for tumor cell survival. Tumor -site Treg cells gain the ability to express VEGFA in and hence play a role in promoting angiogenesis at the tumor microenvironment [23]. Several studies have demonstrated that when cancer advances, the tumor size grows; creating a hypoxic tumor microenvironment that attracts more and more Tregs via the release of a group of chemokines. Due to their immunosuppressive and tumor-promoting properties, these Tregs contribute to the tumor's growth. Tregs promote tumor growth by encouraging endothelial cells to neovascularize in the tumor site so that the quickly multiplying tumor cells may get the nutrients they need. The Treg lineage-specific transcription factor, FOXP3 does not have a binding-site at the VEGFA promoter. Rather, it binds to the promoter of VEGFA with the help of another transcription factor, STAT3, and so promotes its expression in cancer patients and aids in neoangiogenesis [24].
Platelets
According to new findings, platelets are one of the main sources of serum VEGF. Proliferating megakaryocytes secrete VEGF along with hematopoietic cytokines, resulting in autocrine stimulation of their growth, increased endothelial proliferation [25].
Keratinocytes
Recent studies have shown evidence of keratinocytes expressing VEGF during wound healing. The VEGF receptor, on the other hand, -was found to be increased in newly created blood vessels around wound margins. All of these studies point to a paracrine effect of keratinocyte-derived VEGF on angiogenesis.
Autocrine Signaling, VEGF, and Cancer Stem Cells
The effects of VEGF and VEGF receptor-mediated signaling on cancer growth are significant [13]. Many investigations have revealed that VEGF-signaling is essential not just for cancer cells but also for cancer stem cells [26]. There have been numerous debates about the presence or existence of these cells, but it is clear that many tumorous growths contain a small population of self-renewing stem-like cells that can initiate new tumor growths. Studies with transgenic mouse models have revealed that VEGF has a cell-autonomous and angiogenesis-independent function. VEGF was found to mediate an autocrine proliferative loop. Other research involving squamous carcinoma analyses has revealed that autocrine VEGF-signaling affects cancer stem cell functioning directly. The presence of a separate autocrine signaling loop facilitated by the phosphorylation and activation of VEGFR2 on the surface of cancer cells indicates the presence of VEGFA synthesis and activation of the receptor VEGFR2 on cancer cells. During onset of tumor development, cancer stem cells create a niche close to endothelial cells. The VEGF receptor was blocked, and this had an effect on the size of the cancer stem cell pool, as well as their ability to self-renew. The absence of VEGFA expression in tumor cells was found to reduce cancer stem cell proliferation and renewal in studies. These findings, which were found in vivo and in vitro, clearly demonstrate the relevance of autocrine VEGFA signaling in cancer stem cell maintenance and function [26,27].
Neoangiogenesis and Immune-regulation are Two Fundamental Functions of VEGF
VEGF-induced neoangiogenesis is the determining factor of cancer progression
As the primary mediator of tumor-angiogenesis, VEGF plays a significant role in tumor growth. Oncogene expression, a number of growth hormones, and hypoxia all increase VEGF expression. Cancer cells require blood vessels for nutrition and oxygen to proliferate quickly and form a distinct mass. They go through an angiogenic switch as a result of which the cancer cells release VEGF and other growth factors. These help the tumor expand rapidly by forming new blood vessels in and around it. Endothelial cells are naturally attached to a laminin-rich basement membrane and a 1 to 2 cell thick-layer of supportive pericytes in normal conditions. The entire scenario changes during angiogenesis. The connections between nearby pericytes must be weakened, and the basement membrane around them must be deteriorated. Endothelial cells re-enter the cell cycle [28] and begin to synthesize a basement membrane, which aids in the restoration of their capillary-like morphology and cell cycle withdrawal [29]. These newly formed capillaries are also supported by pericytes, which are attracted to the site and aid in their maturation and stabilization. The tumor vasculature that develops under the influence of VEGF is physically and functionally aberrant. Chronic exposure to angiogenic factors that either support basement membrane proteolysis or antagonize endothelial–pericyte interactions in the tumor microenvironment results in the formation of a relatively unstable, highly-permeable network of vessels that do not fully mature but are capable of supplying nutrients to meet the tumor's increasing metabolic demands. Indeed, greater permeability of these vessels is thought to enhance tumor cell extravasation and, eventually, metastasis. Blood vessel forms are also damaged; they have dead ends and are not structured into venules, arterioles, and capillaries, resulting in excessive interstitial pressure. All of these features of tumor vasculature led to hypoxia and increased VEGF production. Because of its important function in the formation of tumor vasculature, VEGF is a suitable candidate for anticancer therapy.
VEGF is strongly associated with tumor immunosuppression

Figure-3: Angiogenesis and VEGF are strongly associated with tumor immunosuppression and targeting VEGF/VEGFR can enhance anti-tumor immunity: Overproduction of VEGF in the TME can lead to suppressed antitumor immunity via two principal mechanisms; 1. Inhibition of effector immune cell (CD4+ T cell, CD8+ T cell, dendritic cell, antigen-presenting cells) function and 2. Recruitment of immunosuppressive cells (Tregs, MDSCs, TAMs) to the TME. Targeting angiogenesis by VEGF blockers may be an effective strategy to increase the efficacy of immunotherapy by improving effector T-cells function whereas decreasing the number and efficacy of immunosuppressive cells in the TME. Combinatorial therapy which targets both VEGF and immune cells has the potential to transform the tolerogenic TME into the immunogenic TME. (Created in Biorender.com)
Suppression of effector immune cell function: Dendritic cells (DC) are antigen-presenting cells with VEGF-binding surface receptors (mostly VEGFR2). This interaction appears to suppress the transcription factor NFkB, which prevents dendritic cell maturation and results in PDL1-induced DC function blockage [38,39]. VEGF also inhibits monocyte development into dendritic cells. Uninterrupted VEGF exposure significantly reduced dendritic cell growth, while increasing the generation of B cells and immature Gr1+ myeloid cells. VEGF also lowers T cell activities by inhibiting the development of progenitor cells into CD4+ and CD8+ lymphocytes [40]. This happens because continuous VEGF expression is linked to NF-kB transcription factor activity suppression in bone marrow progenitor cells. In a finding from by Ohm group, VEGF inhibited the formation of T cells from early hematopoietic progenitor cells, suggesting that VEGF may have an immunosuppressive effect in malignancies [41]. VEGF significantly reduced the cytotoxic activity of T cells generated from peripheral blood samples and activated T cells displayed higher VEGFR2 [42]. According to Ziogas et al. T lymphocytes from ascites related to ovarian cancer produced similar results [43]. In addition to its direct effects on T cells, VEGF can decrease T-cell activity by up-regulating FasL on the endothelium in conjunction with cyclooxygenase [44,45].
PDL1 is expressed in cancer cells and binds to PD1 to suppress CD8+ T cell immunological responses [46]. PD1/PDL1 expression is associated to a poor prognosis in cancer patients [47]. VEGF induces PD1 expression on CD8+ T cells and Tregs, according to studies [48]. PDL1 is also increased in cancer cells after exposure to VEGF [49]. VEGFA released in the TME increases the expression of PD1 and other inhibitory checkpoints involved in CD8+ T cell exhaustion, which can be countered by anti-angiogenic agents that target VEGFA–VEGFR. PDL1, CTLA4, TIM3, and LAG3-mediated signaling lowers effector T cell proliferation and cytotoxic effects on cancer cells [50], while increasing effector T cell anergy and exhaustion [48]. Hypoxia and subsequent angiogenesis also promote PDL1 and CTLA4 overexpression on MDSCs, TAMs, DCs, and cancer cells directly via HIF1 [51]. HIF2 has also been linked to the expression of PDL1 in cancer cells [52]. Tumor-infiltrating effector T cells are anti-tumorigenic that recognize cancer cells and help in their elimination. VEGF-driven angiogenesis, on the other hand, prevents effector T cells from infiltrating the tumor by lowering the expression of endothelial intercellular adhesion molecule-1, lowering their number in the tumor's vicinity and making the TME tolerogenic [53]. Continuous VEGF exposure reduces the number of T cells and lowers the T cell to B cell ratio in lymph nodes and spleen, implying that VEGF is to held responsible for the tumor-associated thymic deficiency [33]. Normal immune activity in cancer patients gradually deteriorates as the thymus fails to replenish the peripheral T-cell pool after prolonged VEGF overexposure [33].
Recruitment of immunosuppressive cells: VEGF-driven angiogenesis promotes the generation of suppressive immune cells such as Tregs, TAMs, and MDSCs, as well as their infiltration into tumor-sites [40,54]. In ovarian and liver cancers, hypoxic tumor microenvironment and VEGF not only increase angiogenesis but also cause the production of CCL28, which promotes the infiltration of CCR10+ Tregs. Infiltrated CCR10+ Tregs also release VEGFA, which promotes neoangiogenesis and distant metastases [56]. Other tolerogenic leukocyte subsets including myeloid-derived suppressor cells and plasmacytoid dendritic cells also produce VEGFA and facilitate tumor angiogenesis [57]. Treg cells can also contribute directly to the VEGFA pool in the TME. Treg cell elimination also reduces VEGFA upregulation in ovarian cancers. As a result, the cancer immune tolerance and angiogenesis processes are intertwined at multiple levels and operate together to promote tumor growth. In tumor microenvironment, VEGF is also critical for Treg induction and maintenance. Tregs are also susceptible to VEGF in some situations because they express VEGFR2 [58]. HIF1 has been linked to Treg differentiation and recruitment to the TME via upregulation of CCL22 and CCL28, in addition to VEGF [59]. During hypoxia, basal-type breast cancer cells increase CXCL12 expression, causing hypoxia-driven Treg recruitment via CXCL12/CXCR4-axis [60]. Thus, therapies that target the CXCL12/CXCR4 and HIF pathways, as well as Treg, may be advantageous for this subset of breast tumors that are less likely to respond to standard chemotherapy. According to reports, the immune evasion mechanism of developing tumors is due to the concentration of these immunosuppressive Tregs in tumor tissue.
By exhibiting many phenotypic characteristics, macrophages often behave as immunosuppressive cells in the tumor microenvironment [61]. M1 macrophages are inflammatory macrophages that aid in the phagocytosis and antigen identification of cancer cells. Tumor growth factors and anti-inflammatory cytokines are produced by M2 macrophages to suppress the host immune response, resulting in tumor progression [61]. TAMs are M2 macrophages that play a key role in immune evasion and are recruited into the TME by tumor-secreted VEGF [62]. Infiltrated TAMs release cytokines and enzymes, such as matrix metalloproteinase-9, that promote neoangiogenesis and degrade extracellular matrix for efficient tissue invasion and metastasis [63]. TAM's interaction with tumor cells has been demonstrated to result in increased VEGFA production, consequent angiogenesis, and tumor aggressiveness. Similarly, hypoxic-TME and subsequent HIF1α and VEGF expression play a role in the polarization of M1 to M2 macrophage phenotype and differentiation of TAMs from immature myeloid cells [64,65].
MDSCs are a type of myeloid cell that can differentiate into other suppressive immune cells and are important in tumor immunosuppression. MDSCs are activated by VEGF released by tumors and can increase angiogenesis directly by secreting more VEGF [66]. MDSC increase in the TME is significantly linked to tumors that secrete a lot of VEGFs. HIF1α promotes MDSC activity and differentiation in the hypoxic-TME, as well as MDSC differentiation into TAMs [67].
Therapeutics
Clinical trials targeting VEGF were initiated in the late 1990s based on the critical role of VEGF in cancer progression, with the first success in 2005 in patients with front-line metastatic breast cancer. This proved the advantages of combining the chemotherapeutic drug paclitaxel with the monoclonal anti-VEGF antibody bevacizumab. Based upon this success, numerous anti-VEGF agents, including drugs that suppress VEGFR2, are now being explored in cancer patients, either alone or in combination with conventional therapy regimens.
Targeting neovasculature
Physical compression, as well as abnormal angiogenesis, can result in irregular vessel formation and impaired blood circulation in tumor tissue [68]. Abnormal blood vessel formation also leads to a decrease in the infiltration of effector immune cells, which mediate immune escape and can reduce the efficacy of immunotherapy by impeding drug delivery. Furthermore, aberrant vasculature promotes the Warburg effect, which accumulates lactic acid within the TME, which is another important cause of suppressed anticancer immunity [36,68]. Thus, targeting VEGF/VEGFR to improve vascular dysfunction - a process known as vascular normalization - not only inhibits the sprouting of new vessels [69], but it may also normalize the vasculature and improve the delivery and efficacy of anticancer drugs, improve oxygen levels, and overcome immunosuppression [70]. Several investigations have established that normalization of tumor vasculature leads to increased effector T-cell infiltration, particularly CD8+ T cells [71-73]. T cell extravasation into the TME is influenced by the levels of expression and clustering patterns of intracellular adhesion molecule-1 (ICAM1) and vascular cell adhesion molecule-1 (VCAM1) [69]. VEGF inhibits leukocyte-endothelial interactions by down-regulating ICAM1 and VCAM1 expression and clustering [74]. A VEGF antibody or inhibitor can reverse this [69]. In turn, aberrant vasculature can be cured by immune cells, as demonstrated in a recent study in experimental breast tumor models in which effector CD4+ T cells were found to both restore tumor vasculature and decrease hypoxia [75]. Similarly, anti-PD1 or anti-CTLA4 therapy promoted vascular perfusion in breast and colon tumor models by promoting CD8+ T-cell accumulation and IFNg production, implying that better vasculature is dependent on increased T-cell mediated immunity [76]. These findings suggest that vascular and T-cell function in cancer are mutually controlled processes that can be treated by either.
Targeting VEGF/VEGFR to enhance anti-tumor immunity
Immunologically, cold tumors conceal their tumor antigens, making them poor targets for immune checkpoint inhibitors. Anti-angiogenic therapy that targets VEGF signaling can convert cold tumors into hot tumors with a favorable microenvironment for cell targeting [77]. As a result, the most effective therapeutic strategy is a combination of an immune checkpoint inhibitor and VEGF-targeted therapy.
Targeting VEGF/VEGFR to enhance effector T-cell function by antiangiogenic agents: VEGF is a crucial link between tumor growth and the immune response directed at that tumor since it promotes both neoangiogenesis and immunosuppression [78]. As a result, targeting VEGF may be an effective strategy for improving immunotherapy efficacy. Some clinical and experimental studies support the hypothesis that treating cancer patients with VEGF blockers improves anti-tumor immunity (Figure 3). For example, in patients with metastatic breast cancer, bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody, has been found to regulate the vasculature, restore DC maturation, and diminish Tregs [75,78]. Both B cell and T cell counts increase in patients receiving bevacizumab-based first-line therapy for metastatic colorectal cancer [79,80]. Sunitinib is a tyrosine kinase inhibitor with many targets that can inhibit VEGFR1, VEGFR2, and VEGFR3. In 2006, the FDA approved sunitinib for the treatment of renal cell carcinoma (RCC) and imatinib-resistant gastrointestinal stromal cancer (GIST) [48,81]. Sunitinib was reported to lower the expression of IL10, FOXP3, PD1, CTLA4, and BRAF in isolated tumor-infiltrating lymphocytes (TILs) but raise Th1 cytokine (IFNg) in an MCA26 (colon cancer cells) harboring animal model. TILs from sunitinib-treated animals had an enhanced proportion and cytotoxic function of CD4+ and CD8+ T cells, whereas expressions of the checkpoint molecules PD1 and CTLA4 were reduced. These findings suggest that sunitinib can affect the TME, altering cytokine and co-stimulatory molecule expression profiles that may promote T-cell activation and Th1 response [82]. Similarly, Schmittnaegel et al. found that inhibiting both angiopoietin-2 and VEGFA might induce an anticancer immune response by raising the proportion of cytotoxic CD8+ T cells in both transgenic and transplanted mammary tumor models [83]. Except for the spleen, all lymphoid organs have high-endothelial venules (HEVs) [84] and specialized post-capillary venules with portals via which blood-borne lymphocytes reach secondary lymphoid organs [85,86]. Recent research has suggested that HEVs can form in tumors and that their existence is related with a smaller tumor size and a better patient outcome. Allen et al. recently revealed that combining anti-VEGFR2 and anti-PDL1 antibodies could produce HEVs in animal models. These HEVs increase effector T cell infiltration and activity by activating lymphotoxin receptor (LTR) signaling, thereby improving therapeutic efficacy [85]. Thus, by targeting VEGFR2, HEVs can serve as a powerful mediator to increase T cell infiltration.
Targeting VEGF/VEGFR to decrease the number and function of immunosuppressive cells: Overexpression of VEGF in the TME is associated with intratumoral Tregs, which is associated with a poor prognosis [59]. According to Suzuki et al., FOXP3hi Tregs mostly express VEGFR2, whereas FOXP3lo Tregs do not [87]. Neuropilin, an immunosuppressive surface protein expressed by Tregs that act as VEGF co-receptors, enhances VEGF binding affinity for VEGFRs [88]. The combination of these two signaling pathways may increase Treg activation and result in a tolerogenic TME [78]. In several neoplasias, rationally targeting VEGFA/VEGFR can modify anti-tumor immunity by inhibiting the activity of inhibitory Tregs. Sunitinib has also been shown in tumor-bearing mice and patients with metastatic renal carcinoma to lower the number and efficacy of Tregs [89]. It has also been demonstrated that the number of Tregs reduced in patients receiving bevacizumab for metastatic colorectal cancer. Similar to the findings of Suzuki et al. [58], these experiments suggest that VEGF could enhance Treg proliferation, and that this proliferation could be prevented by VEGF/VEGFR2 inhibition. Furthermore, the anti-VEGFR2 antibody DC101 and a VEGFR1/R2 chimeric receptor could diminish the amount of Tregs in cancers [90]. In murine tumor models, VEGF-signaling inhibition can improve the efficacy of GMCSF-secreting tumor cell immunotherapies [91]. The improved anticancer activity is linked to an increase in activated CD4+ and CD8+ tumor-infiltrating effector T cells and a significant decrease in suppressive CD4+CD25hi and CD4+FOXP3+ Tregs cells in the cancer. Overexpression of VEGF, on the other hand, increases the amount of Tregs within the TME. All of these findings supported the concept that inhibiting VEGF-signaling in conjunction with immunotherapy increases the ratio of effector T cells to Tregs in the TME, highlighting a viable approach for improving anti-tumor immunity.
Natural inhibitors of angiogenesis

Figure-4: Natural health products inhibiting VEGF: Targeting angiogenesis by VEGF blockers may be an effective strategy to increase the efficacy of immunotherapy by improving effector T-cells function whereas decreasing the number and efficacy of immunosuppressive cells in the TME. Natural health products obtained from various herbs are proven to perform these functions. (Created in Biorender.com)
- Andrographis paniculata: One such resource comes from the herb Andrographis paniculata, the origin being the Indian subcontinent. It is the source of a well-known andrographolide, that specifically binds to the ATP-binding pocket of VEGFR2 and inhibits its kinase activity [96].
- Artemisia annua: The Chinese Wormwood gives the active compound named, Artemisinin [97]. It efficiently lowers the expression of VEGF from tumor cells as well as it inhibits the activation of the nuclear factor kappa-B (NF-kB), an important activator protein in cancer development and progression.
- Viscum album: Viscum album is also known as European Mistletoe. Various studies have shown that it has anti-cancer and anti-angiogenic properties as it can downregulate the expression of VEGF as well as induce cancer cell death [98].
- Curcuma longa: Curcumin, the most active curcuminoid in turmeric, has been demonstrated in numerous studies to inhibit the growth of cancer cells [99]. It suppresses the transcription of two main angiogenesis factors, VEGF and bFGF, in addition to downregulating the expression of matrix metalloproteinase-2 (MMP2) and upregulating the tissue inhibitor of metalloproteinase-1 (TIMP1) [100,101].
Other natural compounds obtained from various natural sources can likewise have a powerful effect on angiogenesis [102]. Grape seed extract, for example, includes the phytoalexin Resveratrol [103,104]. Resveratrol has the ability to prevent VEGF-induced angiogenesis via disrupting reactive oxygen species-dependent src kinase activation and subsequent VE-cadherin tyrosine phosphorylation [105]. Green tea [106,107] is another popular daily product that is gaining popularity. Tea is high in polyphenols and catechins. These have anti-angiogenic effects [108] because they inhibit VEGF transcription, and this benefit may make them useful in cancer treatment. Many in vitro and in vivo studies are being conducted to explore anti-angiogenic properties in various natural health products. One significant advantage of phytochemicals is that they affect not only on angiogenesis but also on many cell-signaling pathways. However, whether these compounds alone or in combination are required for clinical studies must be established.
What is the current anti-angiogenic situation?
Overall, the advantages of anti-angiogenics on survival have been underwhelming, underscoring the need for more effective therapeutic regimens to be developed. Immune checkpoint inhibition, in particular, has been clinically validated as an effective treatment for a variety of cancers with promising results, and there is significant promise in combining immunotherapy drugs with traditional anti-angiogenics.
Antibodies against T-cell immunological negative regulators (PD1, PDL1, and CTLA4) directly boost the immune system, resulting in extraordinary clinical success in a range of malignancies and FDA approval of the following immune checkpoint targeted immunotherapies [109]. Antiangiogenic therapy enhanced anti-PDL1 treatment by promoting vascular changes such as high endothelial venule development and vessel normalcy, which allow for increased cytotoxic T-cell infiltration and subsequent tumor cell death. Recent research further claimed that immune checkpoint inhibition promoted CD4+ T cell activation, as evidenced by increased pericyte coverage, improved tumor vessel perfusion, and lower vascular permeability, all of which resulted in altered tumor progression. Furthermore, bevacizumab-induced VEGF inhibition improved the antigen-presenting capacity of circulating dendritic cells in CRC patients, indicating a new mechanism for bevacizumab's immunological actions in the context of checkpoint blocking. Combining angiopoietin-2 (Ang-2) suppression with VEGF pathway blockage can boost the beneficial effect on immune response [110,111]. Ang-2, like VEGF, is a crucial factor in angiogenesis. A bispecific antibody that binds both VEGF-A and Ang-2 had a larger impact than single inhibition in various preclinical animals and synergized with PD-1 blocking [83,111].
Multiple trials combining VEGF-targeted treatment with checkpoint inhibitors are currently underway, and the synergy between anti-angiogenics and immunotherapy is clear based on both preclinical and translational data. Melanoma, breast cancer, CRC, RCC, and other malignancies are undergoing clinical studies. A clinical trial in patients with metastatic breast cancer that combined the anti-CTLA4 antibody ipilimumab with the anti-VEGF antibody bevacizumab recently revealed encouraging efficacy, with a median overall survival of more than 2 years [112]. High-grade toxicity was more common than expected for either drug alone, but it was manageable. Surprisingly, the combination increased the number of CD8+ T cells and dendritic cells in the TME, signifying immunotherapeutic effector mechanism synergism and warranting further research. Furthermore, nanotechnology-based strategies may increase anti-angiogenic therapy, pharmacokinetic properties, and tumor accumulation [113]. The humanized tri-specific nanobody BI 836880 was just released, and it contains two single-variable domains that inhibit VEGF and Ang2, as well as an albumin module for half-life extension.
Future Perspective
VEGF is critical for embryonic angiogenesis, although its activity in adults is mostly limited to the angiogenic processes of the female reproductive cycle and wound healing. VEGF, on the other hand, plays a critical role in the pathogenesis of a wide spectrum of human malignancies [114]. During cancer progression, rapidly proliferating cancer cells depend on neoangiogenesis for survival and proliferation [101]. Other tumor-associated cells, such as Tregs, M2-macrophages, and cancer stem cells, contribute to angiogenesis by secreting VEGF in addition to cancer cells [25]. VEGF, in addition to acting as a mitogenic signal for vascular endothelial cells, protects tumor vasculature from apoptosis by inducing anti-apoptotic factors, mediates the activation of extracellular matrix degrading enzymes, and promotes tumor-immune evasion by recruiting immunosuppressive cells such as Tregs, TAMs, and MDSCs. The combination action of VEGF as a pro-angiogenic factor and an immunouppressor renders the TME tolerogenic, allowing tumor growth and spread. As a result, inhibiting VEGF and neoangiogenesis has been the primary focus of researchers today in order to halt the spread of metastatic cancer. VEGF-targeted therapy has the potential to operate as immunomodulators, enhancing immune system functioning [14]. Combinatorial therapy that targets both VEGF-signaling and the immune system is becoming more successful. In in-vivo tumor models, natural inhibitors show a strong therapeutic potential against VEGF-induced neoangiogenesis [95]. However, whether or if blocking VEGF-signaling is a viable method to prevent cancer progression remains unknown [115]. New research is being conducted in order to develop successful combinatorial therapies that target both neoangiogenesis and the immune system. This review attempts to summarize the present state of knowledge about new treatment advances to mitigate the effects of VEGF-induced neoangiogenesis and immunosuppression.
Abbreviations
bFGF: basic Fibroblast Growth Factor; HIF: Hypoxia-Inducible Factor; MMP: Matrix Metalloproteinase; PDGF: Platelet-Derived Growth Factor; VEGF: Vascular Endothelial Growth Factor; VEGFR: VEGF Receptor; Treg: T-regulatory cell; TME: Tumor Microenvironment
Declarations
Competing interests
The authors declare no potential conflicts of interest.
Authors' contributions
SD and TS did the background literature study and prepared the initial draft; GS supervised the entire project and made final corrections to the draft.
Acknowledgment
The study was funded by grants from Department of Biotechnology, Government of India. GS is National Academy of Science (India) Platinum Jubilee Senior Scientist.
References
2. Holmes DI, Zachary I. The vascular endothelial growth factor (VEGF) family: angiogenic factors in health and disease. Genome Biology. 2005 Feb;6(2):1-0.
3. Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69(Suppl. 3):4-10.
4. Azimi-Nezhad M. Vascular endothelial growth factor from embryonic status to cardiovascular pathology. Reports of Biochemistry & Molecular Biology. 2014 Apr;2(2):59.
5. Vempati P, Popel AS, Mac Gabhann F. Extracellular regulation of VEGF: isoforms, proteolysis, and vascular patterning. Cytokine & Growth Factor Reviews. 2014 Feb 1;25(1):1-9.
6. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proceedings of the National Academy of Sciences. 1995 Jun 6;92(12):5510-4.
7. Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. Independent function of two destruction domains in hypoxia-inducible factor-α chains activated by prolyl hydroxylation. The EMBO Journal. 2001 Sep 17;20(18):5197-206.
8. Maxwell PH, Dachs GU, Gleadle JM, Nicholls LG, Harris AL, Stratford IJ, et al. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proceedings of the National Academy of Sciences. 1997 Jul 22;94(15):8104-9.
9. Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, et al. Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998 Jul;394(6692):485-90.
10. Fukumura D, Xu L, Chen Y, Gohongi T, Seed B, Jain RK. Hypoxia and acidosis independently up-regulate vascular endothelial growth factor transcription in brain tumors in vivo. Cancer Research. 2001 Aug 15;61(16):6020-4.
11. Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti-and pro-angiogenic therapies. Genes & Cancer. 2011 Dec;2(12):1097-105.
12. Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nature reviews Molecular Cell Biology. 2016 Oct;17(10):611-25.
13. Foekens JA, Peters HA, Grebenchtchikov N, Look MP, Meijer-van Gelder ME, Geurts-Moespot A, et al. High tumor levels of vascular endothelial growth factor predict poor response to systemic therapy in advanced breast cancer. Cancer Research. 2001 Jul 15;61(14):5407-14.
14. O'Reilly MS. „Holmgren, L., Chen, C, and Folkman, J.(1996). Angiostatin induces and sustains dormancy of human primary tumors in mice. Nat. Med.;2:689-92.
15. Kubota Y. Tumor angiogenesis and anti-angiogenic therapy. The Keio Journal of Medicine. 2012 Jun 25;61(2):47-56.
16. Ramakrishnan S, Anand V, Roy S. Vascular endothelial growth factor signaling in hypoxia and inflammation. Journal of Neuroimmune Pharmacology. 2014 Mar;9(2):142-60.
17. Corliss BA, Azimi MS, Munson JM, Peirce SM, Murfee WL. Macrophages: an inflammatory link between angiogenesis and lymphangiogenesis. Microcirculation. 2016 Feb;23(2):95-121.
18. Venneri MA, Palma MD, Ponzoni M, Pucci F, Scielzo C, Zonari E, et al. Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood, The Journal of the American Society of Hematology. 2007 Jun 15;109(12):5276-85.
19. Kim OH, Kang GH, Noh H, Cha JY, Lee HJ, Yoon JH, et al. Proangiogenic TIE2 (+)/CD31 (+) Macrophages Are the Predominant Population of Tumor-Associated Macrophages Infiltrating Metastatic Lymph Nodes (vol 36, pg 432, 2013).
20. Coffelt SB, Hughes R, Lewis CE. Tumor-associated macrophages: effectors of angiogenesis and tumor progression. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer. 2009 Aug 1;1796(1):11-8.
21. Dhar S, Bose S, Sa G. Immunometabolomics: The metabolic landscape of immune cells in tumor microenvironment. Tumor and Microenvironment. 2018 Jul 1;1(3):72.
22. Sarkar T, Dhar S, Sa G. Tumor-infiltrating T-regulatory cells adapt to altered metabolism to promote tumor-immune escape. Current Research in Immunology. 2021 Jan 1;2:132-41.
23. Kajal K, Bose S, Panda AK, Chakraborty D, Chakraborty S, Pati S, et al. Transcriptional regulation of VEGFA expression in T-regulatory cells from breast cancer patients. Cancer Immunology, Immunotherapy. 2021 Jul;70(7):1877-91.
24. Kopp HG, Rafii S. Thrombopoietic cells and the bone marrow vascular niche. Annals of the New York Academy of Sciences. 2007 Jun;1106(1):175-9.
25. Brown LF, Berse B, Jackman RW, Tognazzi K, Guidi AJ, Dvorak HF, et al. Expression of vascular permeability endothelial growth (factor) and its receptors in breast cancer. Hum. Pathol.;26.
26. Mercurio AM. VEGF/neuropilin signaling in cancer stem cells. International Journal of Molecular Sciences. 2019 Jan 23;20(3):490.
27. Lee S, Chen TT, Barber CL, Jordan MC, Murdock J, Desai S, et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007 Aug 24;130(4):691-703.
28. Eliceiri BP, Cheresh DA. Adhesion events in angiogenesis. Current Opinion in Cell Biology. 2001 Oct 1;13(5):563-8.
29. Kubota Y, Martin K. GR and Lawley, TJ (1988) Role of laminin and basement membrane in the morphological differentiation of human endothelial cells into capillary-like structures. J. Cell Biol.;107(4):1589-98.
30. Sarkar T, Dhar S, Chakraborty D, Pati S, Bose S, Panda AK, et al. FOXP3/HAT1 Axis Controls Treg Infiltration in the Tumor Microenvironment by Inducing CCR4 Expression in Breast Cancer. Frontiers in Immunology. 2022;13.
31. Sarkar T, Sa G. Infiltrating Treg Cells Suppress Anti-Tumor Immunity in Tumor Microenvironment. Curr Res Immunol. 2020;1:11.
32. Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'shea KS, et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996 Apr;380(6573):439-42.
33. Ohm JE, Gabrilovich DI, Sempowski GD. Vascular endothelial growth factor C gene expression is closely related to invasion phenotype in gynaecological tumor cells. Blood. 2003;101:4878-86.
34. Toi M, Hoshina S, Takayanagi T, Tominaga T. Association of vascular endothelial growth factor expression with tumor angiogenesis and with early relapse in primary breast cancer. Japanese Journal of Cancer Research. 1994 Oct;85(10):1045-9.
35. Rahma OE, Hodi FS. The Intersection between Tumor Angiogenesis and Immune SuppressionAntiangiogenesis and Immunotherapy. Clinical Cancer Research. 2019 Sep 15;25(18):5449-57.
36. Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nature Reviews Clinical Oncology. 2018 May;15(5):325-40.
37. Khan KA, Kerbel RS. Improving immunotherapy outcomes with anti-angiogenic treatments and vice versa. Nature Reviews Clinical Oncology. 2018 May;15(5):310-24.
38. Oyama T, Ran S, Ishida T, Nadaf S, Kerr L, Carbone DP, et al. Vascular endothelial growth factor affects dendritic cell maturation through the inhibition of nuclear factor-κB activation in hemopoietic progenitor cells. The Journal of Immunology. 1998 Feb 1;160(3):1224-32.
39. Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, et al. Blockade of B7-H1 improves myeloid dendritic cell–mediated antitumor immunity. Nature Medicine. 2003 May;9(5):562-7.
40. Huang Y, Chen X, Dikov MM, Novitskiy SV, Mosse CA, Yang L, et al. Distinct roles of VEGFR-1 and VEGFR-2 in the aberrant hematopoiesis associated with elevated levels of VEGF. Blood, The Journal of the American Society of Hematology. 2007 Jul 15;110(2):624-31.
41. Gabrilovich D, Ishida T, Oyama T, Ran S, Kravtsov V, Nadaf S, et al. Vascular Endothelial Growth Factor Inhibits the Development of Dendritic Cells and Dramatically Affects the Differentiation of Multiple Hematopoietic Lineages In Vivo: Presented in part at the Keystone Symposium “Cellular and Molecular Biology of Dendritic Cells,” Santa Fe, NM, March 3-9, 1998, and at the annual meeting of the American Association for Cancer Research, March 28-April 1, 1998. Blood, The Journal of the American Society of Hematology. 1998 Dec 1;92(11):4150-66.
42. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. Journal of Experimental Medicine. 2015 Feb 9;212(2):139-48.
43. Gavalas NG, Tsiatas M, Tsitsilonis O, Politi E, Ioannou K, Ziogas AC, et al. VEGF directly suppresses activation of T cells from ascites secondary to ovarian cancer via VEGF receptor type 2. British Journal of Cancer. 2012 Nov;107(11):1869-75.
44. Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nature Medicine. 2014 Jun;20(6):607-15.
45. Yang J, Yan J, Liu B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front Immunol 9: 978.
46. Iwai Y, Okazaki T, Nishimura H, Kawasaki A, Yagita H, Honjo T. Microanatomical localization of PD-1 in human tonsils. Immunology Letters. 2002 Oct 1;83(3):215-20.
47. Xue S, Song G, Yu J. The prognostic significance of PD-L1 expression in patients with glioma: A meta-analysis. Scientific Reports. 2017 Jun 26;7(1):1-8.
48. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. Journal of Experimental Medicine. 2015 Feb 9;212(2):139-48.
49. Xue S, Hu M, Li P, Ma J, Xie L, Teng F, et al. Relationship between expression of PD-L1 and tumor angiogenesis, proliferation, and invasion in glioma. Oncotarget. 2017 Jul 7;8(30):49702.
50. Ohm JE, Carbone DP. VEGF as a mediator of tumor-associated immunodeficiency. Immunologic Research. 2001 Apr;23(2):263-72.
51. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. Journal of Experimental Medicine. 2014 May 5;211(5):781-90.
52. Tatli Dogan H, Kiran M, Bilgin B, Kiliçarslan A, Sendur MA, Yalçin B, et al. Prognostic significance of the programmed death ligand 1 expression in clear cell renal cell carcinoma and correlation with the tumor microenvironment and hypoxia-inducible factor expression. Diagnostic Pathology. 2018 Dec;13(1):1-9.
53. Griffioen AW, Damen CA, Martinotti S, Blijham GH, Groenewegen G. Endothelial intercellular adhesion molecule-1 expression is suppressed in human malignancies: the role of angiogenic factors. Cancer Research. 1996 Mar 1;56(5):1111-7.
54. Wada J, Suzuki H, Fuchino R, Yamasaki A, Nagai S, Yanai K, et al. The contribution of vascular endothelial growth factor to the induction of regulatory T-cells in malignant effusions. Anticancer Research. 2009 Mar 1;29(3):881-8.
55. Varney ML, Johansson SL, Singh RK. Tumour-associated macrophage infiltration, neovascularization and aggressiveness in malignant melanoma: role of monocyte chemotactic protein-1 and vascular endothelial growth factor-A. Melanoma Research. 2005 Oct 1;15(5):417-25.
56. Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and Treg cells. Nature. 2011 Jul;475(7355):226-30.
57. Rivera LB, Bergers G. Intertwined regulation of angiogenesis and immunity by myeloid cells. Trends in Immunology. 2015 Apr 1;36(4):240-9.
58. Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, et al. VEGFA-VEGFR Pathway Blockade Inhibits Tumor-Induced Regulatory T-cell Proliferation in Colorectal CancerVEGF-A Inhibition Prevents Tumor-Induced Treg Proliferation. Cancer Research. 2013 Jan 15;73(2):539-49.
59. Wada J, Yamasaki A, Nagai S, Yanai K, Fuchino K, Kameda C, et al. Regulatory T-cells are possible effect prediction markers of immunotherapy for cancer patients. Anticancer Research. 2008 Jul 1;28(4C):2401-8.
60. Yan M, Jene N, Byrne D, Millar EK, O'Toole SA, McNeil CM, et al. Recruitment of regulatory T cells is correlated with hypoxia-induced CXCR4 expression, and is associated with poor prognosis in basal-like breast cancers. Breast Cancer Research. 2011 Apr;13(2):1-0.
61. Tamura R, Tanaka T, Yamamoto Y, Akasaki Y, Sasaki H. Dual role of macrophage in tumor immunity. Immunotherapy. 2018 Aug;10(10):899-909.
62. Chen JJ, Lin YC, Yao PL, Yuan A, Chen HY, Shun CT, et al. Tumor-associated macrophages: the double-edged sword in cancer progression. Journal of Clinical Oncology. 2005 Feb 10;23(5):953-64.
63. Shapiro SD. Diverse roles of macrophage matrix metalloproteinases in tissue destruction and tumor growth. Thrombosis and Haemostasis. 1999;82(08):846-9.
64. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014 Sep;513(7519):559-63.
65. Ott PA, Hodi FS, Buchbinder EI. Inhibition of immune checkpoints and vascular endothelial growth factor as combination therapy for metastatic melanoma: an overview of rationale, preclinical evidence, and initial clinical data. Frontiers in Oncology. 2015 Sep 22;5:202.
66. Drake CG, Allcock JM. Postoperative angiography and the “slipped” clip. Journal of Neurosurgery. 1973 Dec 1;39(6):683-9.
67. Kumar V, Gabrilovich DI. Hypoxia‐inducible factors in regulation of immune responses in tumour microenvironment. Immunology. 2014 Dec;143(4):512-9.
68. Jain RK. Antiangiogenesis strategies revisited: from starving tumors to alleviating hypoxia. Cancer Cell. 2014 Nov 10;26(5):605-22.
69. Lanitis E, Irving M, Coukos G. Targeting the tumor vasculature to enhance T cell activity. Current Opinion in Immunology. 2015 Apr 1;33:55-63.
70. Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Research. 2004 Jun 1;64(11):3731-6.
71. Huang W, Richards S, Carbone MA, Zhu D, Anholt RR, Ayroles JF, et al. Epistasis dominates the genetic architecture of Drosophila quantitative traits. Proceedings of the National Academy of Sciences. 2012 Sep 25;109(39):15553-9.
72. Eng C, Kim TW, Bendell J, Argilés G, Tebbutt NC, Di Bartolomeo M, et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): a multicentre, open-label, phase 3, randomised, controlled trial. The Lancet Oncology. 2019 Jun 1;20(6):849-61.
73. Fizazi K, Maillard A, Penel N, Baciarello G, Allouache D, Daugaard G, et al. A phase III trial of empiric chemotherapy with cisplatin and gemcitabine or systemic treatment tailored by molecular gene expression analysis in patients with carcinomas of an unknown primary (CUP) site (GEFCAPI 04). Annals of Oncology. 2019 Oct 1;30:v851.
74. Dirkx AE, oude Egbrink MG, Kuijpers MJ, van der Niet ST, Heijnen VV, Steege JC, et al. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Research. 2003 May 1;63(9):2322-9.
75. Tian L, Goldstein A, Wang H, Ching Lo H, Sun Kim IK, Welte T, et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature. 2017 Apr;544(7649):250-4.
76. Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007 Feb;445(7130):936-40.
77. Tamura R, Tanaka T, Akasaki Y, Murayama Y, Yoshida K, Sasaki H. The role of vascular endothelial growth factor in the hypoxic and immunosuppressive tumor microenvironment: perspectives for therapeutic implications. Medical Oncology. 2020 Jan;37(1):1-4.
78. Kandalaft LE, Motz GT, Busch J, Coukos G. Angiogenesis and the tumor vasculature as antitumor immune modulators: the role of vascular endothelial growth factor and endothelin. Cancer Immunology and Immunotherapy. 2010:129-48.
79. Manzoni M, Rovati B, Ronzoni M, Loupakis F, Mariucci S, Ricci V, et al. Immunological effects of bevacizumab-based treatment in metastatic colorectal cancer. Oncology. 2010;79(3-4):187-96.
80. Martino EC, Misso G, Pastina P, Costantini S, Vanni F, Gandolfo C, et al. Immune-modulating effects of bevacizumab in metastatic non-small-cell lung cancer patients. Cell Death Discovery. 2016 Oct 3;2(1):1-8.
81. Le Tourneau C, Raymond E, Faivre S. Sunitinib: a novel tyrosine kinase inhibitor. A brief review of its therapeutic potential in the treatment of renal carcinoma and gastrointestinal stromal tumors (GIST). Therapeutics and Clinical Risk Management. 2007 Jun;3(2):341.
82. Ozao-Choy J, Ma G, Kao J, Wang GX, Meseck M, Sung M, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Research. 2009 Mar 15;69(6):2514-22.
83. Schmittnaegel M, Rigamonti N, Kadioglu E, Cassará A, Wyser Rmili C, Kiialainen A, et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Science Translational Medicine. 2017 Apr 12;9(385):eaak9670.
84. Karakhanova S, Link J, Heinrich M, Shevchenko I, Yang Y, Hassenpflug M, et al. Characterization of myeloid leukocytes and soluble mediators in pancreatic cancer: importance of myeloid-derived suppressor cells. Oncoimmunology. 2015 Apr 3;4(4):e998519.
85. Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, ET AL. Combined antiangiogenic and anti–PD-L1 therapy stimulates tumor immunity through HEV formation. Science Translational Medicine. 2017 Apr 12;9(385):eaak9679.
86. Ager A, May MJ. Understanding high endothelial venules: lessons for cancer immunology. Oncoimmunology. 2015 Jun 3;4(6):e1008791.
87. Suzuki H, Onishi H, Wada J, Yamasaki A, Tanaka H, Nakano K, et al. VEGFR2 is selectively expressed by FOXP3high CD4+ Treg. European Journal of Immunology. 2010 Jan;40(1):197-203.
88. Graziani G, Lacal PM. Neuropilin-1 as therapeutic target for malignant melanoma. Frontiers in Oncology. 2015 Jun 3;5:125.
89. Adotevi O, Pere H, Ravel P, Haicheur N, Badoual C, Merillon N, et al. A decrease of regulatory T cells correlates with overall survival after sunitinib-based antiangiogenic therapy in metastatic renal cancer patients. Journal of Immunotherapy. 2010 Nov 1;33(9):991-8.
90. Secondini C, Coquoz O, Spagnuolo L, Spinetti T, Peyvandi S, Ciarloni L, et al. Arginase inhibition suppresses lung metastasis in the 4T1 breast cancer model independently of the immunomodulatory and anti-metastatic effects of VEGFR-2 blockade. Oncoimmunology. 2017 Jun 3;6(6):e1316437.
91. Li B, Lalani AS, Harding TC, Luan B, Koprivnikar K, Huan Tu G, et al. Vascular endothelial growth factor blockade reduces intratumoral regulatory T cells and enhances the efficacy of a GM-CSF–secreting cancer immunotherapy. Clinical Cancer Research. 2006 Nov 15;12(22):6808-16.
92. Elice F, Rodeghiero F. Side effects of anti-angiogenic drugs. Thrombosis Research. 2012 Apr 1;129:S50-3.
93. Ingber D. Fujita T, Kishimoto S, Sudo K, Kanamaru T, Brem H. Folkman J: Synthetic analogues of fumagillin that inhibit angiogenesis and suppress tumour growth. Nature. 1990;348:555-7.
94. Lu K, Bhat M, Basu S. Plants and their active compounds: natural molecules to target angiogenesis. Angiogenesis. 2016 Jul;19(3):287-95.
95. C Recio M, Andujar I, L Rios J. Anti-inflammatory agents from plants: progress and potential. Current Medicinal Chemistry. 2012 May 1;19(14):2088-103.
96. Kajal K, Panda AK, Bhat J, Chakraborty D, Bose S, Bhattacharjee P, et al. Andrographolide binds to ATP-binding pocket of VEGFR2 to impede VEGFA-mediated tumor-angiogenesis. Scientific Reports. 2019 Mar 11;9(1):1-0.
97. Mueller MS, Runyambo N, Wagner I, Borrmann S, Dietz K, Heide L. Randomized controlled trial of a traditional preparation of Artemisia annua L.(Annual Wormwood) in the treatment of malaria. Transactions of the Royal Society of Tropical Medicine and Hygiene. 2004 May 1;98(5):318-21.
98. Harmsma M, Grommé M, Ummelen M, Dignef W, Tusenius KJ, Ramaekers F. Differential effects of Viscum album extract Iscador® Qu on cell cycle progression and apoptosis in cancer cells. International Journal of Oncology. 2004 Dec 1;25(6):1521-9.
99. Sa G, Das T. Anti cancer effects of curcumin: cycle of life and death. Cell Division. 2008 Dec;3(1):1-4.
100. Kim JH, Shim JS, Lee SK, Kim KW, Rha SY, Chung HC, et al. Microarray‐based analysis of anti‐angiogenic activity of demethoxycurcumin on human umbilical vein endothelial cells: Crucial involvement of the down‐regulation of matrix metalloproteinase. Japanese Journal of Cancer Research. 2002 Dec;93(12):1378-85.
101. Narayan S. Curcumin, a multi-functional chemopreventive agent, blocks growth of colon cancer cells by targeting β-catenin-mediated transactivation and cell–cell adhesion pathways. Journal of Molecular Histology. 2004 Mar;35(3):301-7.
102. Samant RS, Shevde LA. Recent advances in anti-angiogenic therapy of cancer. Oncotarget. 2011 Mar;2(3):122.
103. CAO Y, ZHAO-DI FU FW, LIU HY, HAN R. Anti-angiogenic activity of resveratrol, a natural compound from... Journal of Asian Natural Products Research. 2005 Jun;7(3):205-13.
104. Igura K, Ohta T, Kuroda Y, Kaji K. Resveratrol and quercetin inhibit angiogenesis in vitro. Cancer Letters. 2001 Aug 28;171(1):11-6.
105. Lin MT, Yen ML, Lin CY, Kuo ML. Inhibition of vascular endothelial growth factor-induced angiogenesis by resveratrol through interruption of Src-dependent vascular endothelial cadherin tyrosine phosphorylation. Molecular Pharmacology. 2003 Nov 1;64(5):1029-36.
106. Sartippour MR, Shao ZM, Heber D, Beatty P, Zhang L, Liu C, et al. Green tea inhibits vascular endothelial growth factor (VEGF) induction in human breast cancer cells. The Journal of Nutrition. 2002 Aug 1;132(8):2307-11.
107. Cao Y, Cao R. Angiogenesis inhibited by drinking tea. Nature. 1999 Apr;398(6726):381-.
108. Lee MJ, Maliakal P, Chen L, Meng X, Bondoc FY, Prabhu S, et al. Pharmacokinetics of tea catechins after ingestion of green tea and (−)-epigallocatechin-3-gallate by humans: formation of different metabolites and individual variability. Cancer Epidemiology Biomarkers & Prevention. 2002 Oct;11(10):1025-32.
109. Osada T, Chong G, Tansik R, Hong T, Spector N, Kumar R, et al. The effect of anti-VEGF therapy on immature myeloid cell and dendritic cells in cancer patients. Cancer Immunology, Immunotherapy. 2008 Aug;57(8):1115-24.
110. Baker LC, Boult JK, Thomas M, Koehler A, Nayak T, Tessier J, et al. Acute tumour response to a bispecific Ang-2-VEGF-A antibody: insights from multiparametric MRI and gene expression profiling. British Journal of Cancer. 2016 Sep;115(6):691-702.
111. Kloepper J, Riedemann L, Amoozgar Z, Seano G, Susek K, Yu V, et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proceedings of the National Academy of Sciences. 2016 Apr 19;113(16):4476-81.
112. Hodi FS, Lawrence D, Lezcano C, Wu X, Zhou J, Sasada T, et al. Bevacizumab plus Ipilimumab in Patients with Metastatic MelanomaBevacizumab plus Ipilimumab. Cancer Immunology Research. 2014 Jul 1;2(7):632-42.
113. El‐Kenawi AE, El‐Remessy AB. Angiogenesis inhibitors in cancer therapy: mechanistic perspective on classification and treatment rationales. British Journal of Pharmacology. 2013 Oct;170(4):712-29.
114. Al Kawas H, Saaid I, Jank P, Westhoff CC, Denkert C, Pross T, et al. How VEGF-A and its splice variants affect breast cancer development–clinical implications. Cellular Oncology. 2022 Mar 18:1-3.
115. Eguchi R, Kawabe JI, Wakabayashi I. VEGF-Independent Angiogenic Factors: Beyond VEGF/VEGFR2 Signaling. Journal of Vascular Research. 2022 Feb 11:1-2.